阿尔茨海默病(AD)是老年人群常见的神经系统变性疾病,临床表现为认知和记忆功能不断恶化、日常生活能力进行性减退、合并各种神经精神症状和行为障碍。特征性病理表现为老年斑形成及神经原纤维缠结。AD病因和发病机制复杂,目前认为遗传和环境因素是促使AD发生的重要因素。AD的传统治疗包括了胆碱酯酶抑制剂、谷氨酸亚型N?甲基?D?天冬氨酸(NMDA)受体拮抗剂、抗β?淀粉样蛋白(Aβ)药物、神经生长因子、非甾体类抗炎药物等。这些治疗能相对减轻AD的症状,但不能阻止病情的进展,因此疗效有限。近年来的研究肯定了金属物质如铜、锌、锰、铁等代谢异常,引发的神经细胞过氧化是导致AD发生的高危因素之一〔1〕,其中铜扮演了重要角色。近年来对铜代谢和AD发病机制的相关研究取得了可喜成果,本文作一简要综述。
1 铜代谢及生理功能
铜是人体必需的微量元素,正常人体内含铜量100~200 mg。在人体内铜具有多种重要生理功能:①参与造血和铁的代谢,影响铁的吸收和储存;②构成许多含铜酶及含铜生物活性蛋白质;③与DNA结合,与维持核酸结构的稳定性有关;④许多酶类都需要一定的铜才能发挥其功能。如在呼吸链中的关键酶?细胞色素C氧化酶中铜是重要的成分之一;参与组成儿茶酚胺生物合成途径的限速酶多巴胺羟化酶;Cu/Zn超氧化物歧化酶(SOD)的辅因子,从而参与清除细胞活性氧,对抗氧化应激。另外、尿酸氧化酶、赖氨酸氧化酶和酪氨酸酶等也都含有铜。人类每日从饮食中摄入0.6~2.0 mg的铜,其中约50%被机体吸收(约0.3~1.0 mg),通过小肠的刷状细胞进入血液中,以离子形式与铜结合蛋白(白蛋白或转铜蛋白)结合,再被肝脏摄取,并在内质网中形成铜蓝蛋白(其活性依赖于与铜的结合),进而运输到肌肉、骨骼及肝脏、皮肤、血液和脑等器官中,其中前两者约占50%~70%。以二价或者一价形式与众多功能蛋白结合发挥其生理作用:使机体内的众多酶和蛋白质保持其功能。在组织细胞内,铜伴侣蛋白还会将金属离子运输到特定的分子中去发挥功能〔2〕。约80%的铜经胆囊排泄,不到4%通过肾脏排除。经胆道排泄入小肠的铜还可以被再摄取利用形成肠肝循环,保持体内铜水平的相对恒定。
2 铜代谢异常与AD法学论文发表
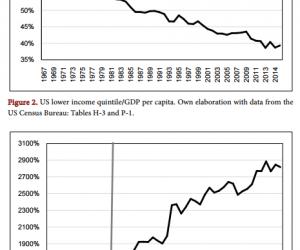
研究表明铜代谢异常与AD有密切关系。铜摄取不足可致神经系统功能失调,大脑功能会发生障碍。其特征表现是大脑皮层广泛的髓鞘脱失和进行性的脊髓脱髓鞘。电镜下发现高尔基体异常,神经元细胞和胶质细胞胞体改变,轴突变性损伤。表现为神经系统活力下降,从而使思维紊乱、反应迟钝,甚至步态不稳、运动失常等。而铜在脑组织中过多聚集引起神经细胞毒性则被认为是导致AD发生的一个重要因素。因为它改变了淀粉样前体蛋白(APP)的正常代谢及参与Aβ的聚集等〔3,4〕。
2.1 铜与APP 代谢 APP是产生Aβ的前体物质,在体内各种组织广泛存在,是一跨膜蛋白质。APP基因约有170 kb,含有18个外显子,位于21号染色体的21q21.2上。APP基因转录后主要形成APP695、APP751、APP770三种同源产物。其中脑组织中主要是APP695,也有APP751和APP770。APP的体内功能目前还不是十分清楚,目前认同的说法是,APP是一种自分泌因子,具有促进细胞增生和神经营养因子的作用,它通过与细胞膜外基质的相互作用介导神经细胞之间的黏附,增加神经突触之间的联系及突触可塑性。APP降解有两个途径,即α途径和β途径,前者先由α?分泌酶(α?secretase) 分解产生较大的N末端片段sAPP?α和跨膜片段C83,C83再经γ?分泌酶作用产生p3和另一胞内APP的C端(APP intracellular domain,AICD)片段,这一途径避免了Aβ的产生,且已有报道sAPPα可对神经细胞产生神经营养和神经保护作用〔5〕;后者先经β分泌酶作用,产生大的N末端片段sAPP?β和小的跨膜片段C 99,后者再经γ分泌酶作用产生Aβ和另一胞内片段AICD,Aβ在细胞内生成后要分泌到细胞外。Aβ是形成淀粉样聚集的主要物质。位于APP124~189位氨基酸残基部位有特异性铜结合位点(CuBD)〔6〕。Kong等〔7〕发现CuBD中两个亲水基团位于其他基团的上方,使金属中心基团形成一种锥体型从而利于二价铜离子的结合并还原。电子顺磁共振和X线吸收光谱学分析其细微结构进一步证实了这种几何关系的存在。有研究显示CuBD主要存在于APP133~189片段上〔8〕。二价铜离子结合于CuBD后被还原为一价铜离子,增加铜的细胞内蓄积,从而增加了神经系统的氧化应激〔9〕。
目前认为铜影响APP细胞培养系统中的物质加工过程,降低Aβ的水平,同时增加APP外结构域分泌。提示铜对APP的调节机制有两方面,作用于APP的合成和Aβ的产生。细胞内APP水平升高的同时伴有Aβ生成的减少。说明Aβ生成减少并不是因为APP分泌总体受抑制,而是铜可以增加APP分泌的同时抑制Aβ的生成。但是机制不十分清楚。法学论文发表
2.2 铜与Aβ代谢 研究表明Aβ寡聚体对培养的神经细胞具有很强的损伤作用〔10〕。Aβ与铜有高度的亲和力,进一步发现铜与Aβ具有双向性,一方面Aβ促使铜离子排出脑外,另一方面铜离子异常使Aβ的毒性增强。最近Streltsov等〔11〕通过X射线方法验证了铜离子与Aβ结合位点的假说,虽然其确切位置尚不清楚,但其促进Aβ聚集及导致氧化应激作用已经被认同〔12〕。有研究表明在饮用水中加入高浓度铜加速了AD模型兔脑组织中Aβ的聚集并引起过氧化事件的发生〔13,14〕。在增加铜的生物利用度抑制了糖原合成酶3β(GSK?3β)后,测定APP转基因模型鼠Aβ含量减少,tau蛋白磷酸化水平明显降低〔15〕。Squitti等〔16〕发现AD患者铜含量明显高于正常对照组,且与肝功相关指标呈负相关性,与直接胆红素呈正相关性。资料表明,脑中毛细血管的低密度脂蛋白受体有清除Aβ的作用,但是过量的铜会对这一作用产生负面影响〔17〕,提示铜参与了Aβ的形成及清除过程。
2.3 铜与载脂蛋白E(ApoE?ε4)代谢 ApoE基因位于第19号染色体长臂上,包括4个外显子和3个内含子,全长3.7 kb。ApoE?ε4等位基因突变是AD的致病因素之一,其表现型和频率存在种族和地域差别。Zappasodi等〔18〕对携带ApoE?ε4 基因的AD患者和未携带ApoE?ε4基因患者进行脑电图(EEG)描记和测定血清铜水平。与对照组比较结果显示,AD组血清铜水平显著高于正常对照组,且与EEG的α波活动呈负相关、与δ波活动呈正相关;进一步分析发现ApoE?ε4基因携带者血清铜水平升高、α活动强于非携带者,氧化作用在δ波活跃的AD中明显增强,提示铜的氧化应激损害可能是参与ApoE?ε4引发AD的一种机制。
3 铜螯合剂治疗AD
上述发现提示通过改善体内铜的内环境,有可能达到治疗AD的目的。因此,金属螯合剂就成为治疗AD的新途径。金属螯合剂可以明显减轻转基因小鼠脑中Aβ的负荷。氯碘羟喹(CQ)是一种铜锌螯合剂,很早就被试用于AD的治疗。Cherny〔19〕等在PAA2576转基因鼠中应用氯碘羟喹后,发现Aβ比对照组减少了49%。最近Ferrada等〔20〕发现脑中的Cu(Ⅱ) CQ络合物影响了血清铜浓度,从而有利于Aβ的降解。关于CQ的机制目前是比较肯定的:在大鼠,CQ进入脑组织结合铜,降低铜所致的过氧化作用从而减少Aβ的聚集〔21〕。在细胞培养中,CQ?Cu 络合物通过增加基质金属蛋白酶?2 (MMP?2)和基质金属白酶?3(MMP?3)显著抑制了Aβ的分泌〔22〕。研究表明Cu(Ⅱ) CQ络合物能使细胞外Aβ40和Aβ42的浓度迅速降低,其机制是Cu(Ⅱ) CQ络合物刺激磷脂酰肌醇3?激酶( phosphatidylinositol 3 kinase,PI?3K)通路导致下游的丝氨酸苏氨酸蛋白激酶(Akt)磷酸化和抑制糖原合酶激酶?3β(GSK?3β),进一步通过C?Jun氨基末端激酶(JNK)的活动和细胞外信号调节激酶(ERK)磷酸化引起MMP?2和 MMP?3的增加,MMP?2及MMP?3是降解Aβ的酶类〔4,22〕。在动物实验取得良好结果后,CQ正式进入临床试验阶段。在澳大利亚进行的CQ的二期临床试验中,36名AD患者被随机分组,用CQ治疗组与使用安慰剂的对照组比较,认知能力明显得到改善,且血浆Aβ42水平也显著降低,同时并没有引起以往曾经出现过的亚急性视神经脊髓炎〔23〕。最近,在临床试验中二代的CQ的8?OH喹啉衍生物被证明具有更强的穿透血脑屏障的作用,共有78名早期AD患者作为受试者参与了实验。在为期12 w的试验中,50 mg和250 mg两种药物剂量都被证明安全且有效,并且在用250 mg的药物剂量时,患者脑脊液(CSF)中的Aβ浓度更是显著降低。另外,Malm等人〔24〕利用另一种铜离子络合pyrrolidine dithiocarbamate (PDTC)治疗转基因AD小鼠(APP/PS1),发现小鼠认知功能明显改善。这些研究均提示了应用铜螯合剂治疗AD是有效的。
4 问题与展望法学论文发表
虽然AD的病因复杂,发病机制尚不十分清楚,现有药物的疗效不甚理想,但随着铜代谢与AD发病机制相关性研究的逐步深入,金属螯合剂治疗AD将成为新的可能途径之一。目前尚待突破的难题是找到一种特异性好的,并且用什么方法给予患者什么样的浓度能够最大限度地发挥其治疗作用而降低其负面作用。








论文发表 是一个专门从事期刊推广、论文发表、论文发表辅导的机构。融合收集数百家期刊杂志社征稿评职称,发表论文,以供广大作者免费阅览,以期待各位作者在短时间内掌握每种期刊征稿要求、审稿范围,在日常繁重的工作中短时间开心、放心快速投稿、在适合的时间拿到刊物,评上职称,不为挤不上评职称的末班车而烦恼。
本站主要整合如下评职称,发表论文:
教育论文发表
经济论文发表
科技论文发表
医学论文发表
计算机论文发表
文学论文发表
农业论文发表
学报论文发表
其它论文发表等,另期待更多的杂志编辑与本站联系文:
85782530(普刊发表)
82534308(核心发表)共同以正期刊界正刊之气,维护学术良好的氛围。
免责声明:本网所提供的信息资源如有侵权、违规,请及时告知!
论文网版权所有, 本站提供论文发表 论文投稿 发表论文 发表文章
文章只代表作者观点,并不意味着本站认同,部分作品系转载,版权归原作者或相应的机构;若某篇作品侵犯您的权利,请来信告知:lunww@126.com
中国论文网全权所属国际域名:www.lunww.com 中文域名:论文发表 技术支持:论文发表 国家双软认定单位 法律顾问:全民安律师事务所
期刊合作加盟咨询:5715378(加盟、投诉、建议) 论文发表咨询电话:18262951856 论文发表投稿邮箱:lunww@126.com
Powered by 论文发表 © 2010-2020

苏ICP备19023845号-4