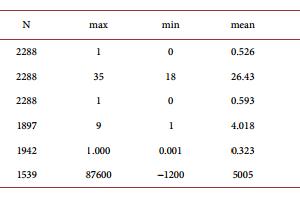
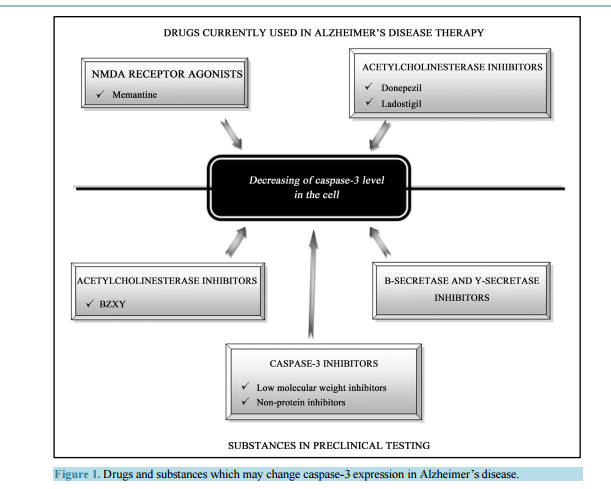
 Activated Caspase-3 and Neurodegeneration and Synaptic Plasticity in Alzheimer’s Disease Anna Płóciennik, Michał Prendecki, Ewelina Zuba, Marcin Siudzinski, Jolanta Dorszewska Laboratory of Neurobiology, Department of Neurology, Poznan University of Medical Sciences, Poznań, Poland Email: dorszewskaj@yahoo.com Received 19 June 2015; accepted 25 August 2015; published 28 August 2015 Copyright © 2015 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract In Alzheimer’s disease (AD) there is a loss of neurons in different areas of the brain, including hippocampus, temporal and frontal cortex. Molecular mechanism of neuronal death in AD is not clear. It seems that apoptosis is a major form of neuronal cell death in this neurodegenerative disease. Neurons in both experimental animals and AD brains showed apoptotic changes, including DNA fragmentation, microRNA expression and caspase-3 activation. Activated caspase-3 may lead to amyloid precursor protein (APP) cleavage, extracellular senile plaques (SPs) and intracellular neurofibrillary tangles (NFTs) formation and neuronal loss, and it contributes to synaptic plasticity and cognitive dysfunction, especially in early stage of AD. Moreover, changes of caspase-3 activity were observed after therapy with memantine (NMDA receptor antagonist), donepezil and ladostigil (acetylcholinesterase inhibitors) in AD patients. It has been shown that both mementine and acetylcholinesterase inhibitors protect neurons from the caspase-3 activation. It seems that caspase-3 may be a potential diagnostic factor and target for pharmacotherapy of AD patients. Keywords Caspase-3, MicroRNA, Apoptosis, Synaptic Plasticity, Experimental Models, AD 1. Introduction Alzheimer’s disease (AD) is one of the most common neurodegenerative disorders of human beings. The disease manifests usually after the age of 65 years. The frequency of AD in this age group is 1% - 3% and doubles every five years, exceeding 40% in 85-year-old group. It is estimated that the number of patients suffering from A. Płóciennik et al. 64 AD is 21 - 24 million and this number will double in next 15 years. The disease occurs approximately two times more often in female than male [1]. AD usually lasts approximately for 10 years (less in men more for females). During the first 10 - 20 years the disease may be asymptomatic or manifest as mild cognitive impairment [2]. Then there is a growing mental process of memory and cognitive function may also experience changes in personality, delusions, confusion and depression that impair and ultimately prevent the independent functioning of the patient. Patients in last stage of the disease gradually lose the ability of communication, navigation, swallowing, etc. Patients become completely dependent on relatives and caregivers and require a constant supervision. The disease is usually fatal because of extracerebral complications [3]. There are two forms of AD, sporadic (SAD) with non-Mendelian genetic pattern and autosomaly dominantly inherited familial AD (FAD). Mutations of the amyloid precursor protein (APP), the presenilin 1 (PS1) and the presenilin 2 (PS2) coding genes (APP, PSEN1 and PSEN2, respectively) have been linked to FAD. Approximately 95% of AD cases occur as a SAD form, and the rest have autosomal dominant inheritance (FAD) [4] [5]. SAD was shown to be a multifactorial disease, in 70% determined by genetic factors and in 30% determined by non-genetic factors, e.g. environmental factors [6]. The most prominent genetic factors may be accounted genes involved in cholesterol metabolism and tricking, e.g. ABCA1, ABCA7, APOE and SORL1 [7] [8]. Of these genes APOE seems to be the most significant, where APOE ε4 allele is responsible for approximately 1/4 of SAD cases. It was also demonstrated that APOE ε4 may augment formation of senile plaques and impair clearance of AD pathological proteins [9]. The AD has been also associated with rare variants in other genes e.g. CR1, EPHA1, HLA, INPP5D, MS4A, TREM2 and TREM2L, responsible for immunology processes [10] and TP53-regulating apoptosis [11]. The most recent data indicate that AD may be also associated with dysregulation in microRNA expression [7] [12]. The most important non-genetic risk factors of AD include: being female, age over 65 years, low level of education, a poor nutritional status, a presence of relatives suffering from AD, an aluminum intoxication, chronic use of benzodiazepines, a tobacco use, a head trauma with loss of consciousness, a brain ischemia and infection of the central nervous system [13]. The most vulnerable areas in AD brains are the hippocampus and temporal, frontal, entorhinal cortex, and amygdale [14]. Neuropathological hallmarks of AD are neuronal loss, extracellular senile plaques (SPs), containing β-amyloid (Aβ) and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau proteins. It is known that Aβ accumulated in SPs is neurotoxic [15]. The Aβ is produced in brain in the manner of proteolysis. In physiological conditions—90% of APP, a transmembrane neuronal protein is cleaved by two enzymes: α- and γ-secretases, whose active subunits are coded by ADAM10, PSEN1 and PSEN2 genes, respectively. This leads to production of non-toxic proteolysis products [16]. In AD the second mechanism of APP lysis by β- and γ-secretases is activated, leading to formation of 42 aminoacid, highly hydrophobic Aβ. This peptide undergoes quick oligomerization and if the clearance mechanisms are disturbed (e.g. due to rare variants in respective genes), the oligomers bind to form larger structures—neurotoxic SPs [17]. Hyperphosphorylation of tau protein results in paired helical filaments (PHFs) formation which agglomerates in pericarions and creates NFTs which lead to disruption of intracellular transport [18]. Furthermore, NFTsbearing neurons have been suggested to be responsible for neuronal death in AD [19]. Data in literature indicated that Aβ and NFTs induce oxidative stress in neurons, and thus may be associated with neuronal loss in AD [18] [19]. Another neuropathological lesion in AD is granulovacuolar degeneration [20]. Up till now, the cause of damage to neurons and memory loss in the pathogenesis of AD is not clearly explained. One possible cause of neuronal loss in AD appears to be increased process of apoptosis [21] [22]. 2. The Role of Activated Caspase-3 in Pathogenesis of Alzheimer’s Disease Several studies [21]-[23] showed that apoptosis leads to neuronal death in AD. Changed expression of proteins linked to apoptosis, such as Bad, Bcl-2, Bax, p53 and Fas was reported in AD brains [24] [25]. Caspase-3, a protease, is thought to be an essential executioner caspase in the apoptosis [26]. Forming of active caspase-3 performs an irreversible step of the apoptotic pathway. Numerous studies have presented elevated level of caspase-3 in AD [27]-[30]. Su et al. [27] showed elevated active caspase-3 expression within SPs in postmortem hippocampus, frontal and entorhinal cortex of AD brains. Another studies showed that caspase-3 is prominent caspase implicated in A. Płóciennik et al. 65 APP cleavage [31]. Moreover, Gervais et al. [31] indicated that activated caspase-3 may lead to SPs formation, as well as it may play an essential role in neuronal loss in AD. The study of Khan et al. [4] showed elevated level of Aβ associated with increased caspase-3 activation in cytoplasmic hybrid cells created from mitochondrial DNA of SAD patients. Moreover, in this study secretion of Aβ was lowered by caspase-3 inhibition by incubation it with an antioxidant, pramipexol. According to the authors, mitochondrial genes in SAD may be responsible for Aβ oversecretion mediated by casapse-3 [4]. However, the study of Gastard et al. [28] indicated an increased expression of caspase-3 immunoreactivity in postmortem hippocampus, entorhinal cortex and cornu amonis of AD brains. In this study expression of activated caspase-3 was significantly higher in early stage of AD and it was colocalised with PHF-bearing neurons. Moreover, activated caspase-3 was lowered in advanced AD and in late stage of this disease was colocalised with classic NFTs. Su et al. [27] also demonstrated elevated levels of activated casapse-3 in postmortem hippocampus, frontal and etorhinal cortex and a prominent colocalisation between active casapse-3 and NFT-bearing neurons (pre-tangle neurons, early stage NFTs and mature NFTs). Another study showed that caspase-3 may be implicated in tau protein phosphorylation as well [32]. It seems that caspase-3 may be mediated NFTs formation and neuronal death in AD. Caspase-3 immunoreactivity was also reported in granules of granulovacuolar degeneration (GVD) in AD [27]-[29]. Granulovacuoles are considered as autophagic in origin [33] [34]. Activated caspase-3 in GVD may indicate the presence of self-repair processes in neurons. An existence of active caspase-3 in GVD might be elucidated by a lysosomal activation of caspases [35] [36], involving an activation of apoptosis which may lead to cell death and neuronal loss in AD. This notion is supported by evidence that apoptosis and autophagy are related processes and that there are autophagic forms of apoptotic pathway [37] [38]. Kovacs et al. [30] showed elevated caspase-3 level on the human H4 neuroglioma cell line, expressing mutation in PSEN1 gene. Increased level of caspase-3 was a result of staurosporine (STS) treatment, which has a proapoptotic effect. Authors showed that caspase-3 activation leads to cleaving of PS1. In the study of Tesco et al. [39] also showed that caspase may induce cleavage of PS1 and interrupts the interaction of PS1 with β-catenin which may contribute to cell death. 3. Activated Caspase-3 in the Experimental Models of Alzheimer’s Disease Currently pathogenesis of AD can be more clearly explained using experimental models of AD. Transgenic mouse models are very often used in the study of many neurodegenerative diseases, such as AD, Parkinson’s or Huntington’s diseases. The animal models carry mutations in specific genes which are involved in the pathogenesis of neurodegenerative diseases [40]-[46]. It is known that casapse-3 plays a significant role in the apoptosis and its level is markedly increased in diseases where such phenomenon occurs e.g. AD. The elevated level of casapse-3 was observed in hPS2m-Tg mice (carrying the human mutant PS2) and hPS2w-Tg mice (carrying the human wild PS2) compare to non-Tg mice [42]. Also, the increase in casapse-3 level was observed in a Tg2576 transgenic mouse model, which harbors the human APPswe mutant allele linked to FAD [45]. In this mouse model a significant increase in caspase-3 activity was detected in Tg2576 mice at the age 3 months (young adult), but not at 2 months. In another mouse model of AD, transgenic PS/APP, which expressed both the Swedish double mutations of APP (K670N/M671L) and PS1 (PS1M146L), a markedly elevated level of caspase-3 was observed as well [46]. In AD one of the major clinical features is memory impairment. The hippocampus plays a crucial role in memory processes, thus an increased caspase-3 activity should be detected in this particular structure. Indeed, in each quoted transgenic mouse model an enhanced caspase-3 level was disclosed in the hippocampus. In hPS2m-Tg and hPS2w-Tg mice it was observed in hippocampus-dentate gyrus but also in the cerebral cortex [42]. In Tg2576 transgenic mouse model an increase of caspase-3 was detected in CA1 pyramidal neurons [45]. In PS/APP transgenic model [APP (K670N/M671L) and PSEN1 (PS1M146L)] the elevation of caspase-3 activity was disclosed not only in the hippocampus (especially in the stratum lucidum of the hippocampal CA3 area) but also in other brain regions, including cortex, striatum, thalamus, and brainstem [46]. Also at the ultrastructural level there have been reported morphological changes typical for the apoptosis. Indeed, adult PS/APP mice exhibited pyknotic caspase-3-positive neurons which revealed such apoptotic changes as DNA fragmentation, caspase-3 activation, caspase-cleaved spectrin generation, chromatin margination and condensation, chromatin balls, and nuclear membrane fragmentation. What is more, the majority of caspase-3- A. Płóciennik et al. 66 immunoreactive neurons were grossly shrunken or fragmented without recognizable processes. Notably, activated caspase-3 and caspase-3-cleaved spectrin were abundant in autophagic vacuoles, accumulating in dystrophic neuritis of PS/APP mice [46]. Because synapses are necessary in the transmission of neuronal impulses which are involved in memory processes, it is supposed that these neuronal structures should be particularly affected in AD, thus presenting a high level of casapse-3 activation. In Tg2576 mice caspase-3 was more abundant in the synaptosomal fraction than in total homogenate [45]. Caspase-3 immunoreactivity was found in dendritic spines and it was more abundant in Tg2576 neurons than in their wild-type counterparts. In the spine, the immunoprecipitates were specifically concentrated in the postsynaptic sites. No immunoreactivity however was disclosed in cell soma, nuclei, presynaptic terminals or surrounding glial projections in Tg2576 mice. The study of D’Amelio et al. [45] showed that active caspase-3 is involved in the removal of the GluR1 subunit of AMPA-type receptor from postsynaptic sites which may lead to alterations of glutamatergic synaptic transmission and plasticity and correlated with spine degeneration and a deficit in hippocampal-dependent memory. The study revealed that these molecular modifications of glutamatergic synaptic transmission triggered by an active caspase-3 were performed by the activation of calcineurin A. In vivo, an elevation of caspase-3 activity in the hippocampus of the 3-month-old Tg2576 was associated with an increase of both activity and processing of calcineurin when compared with the wild type mice. Calcineurin A is proteolytically activated by caspase-3 and is responsible for Aβ-mediated synaptic removal of AMPAR which leads to the loss of dendritic spines [45]. In Tg2576 mice the caspase-3 activity increase was matched by a deficit in hippocampal-dependent contextual fear conditioning (CFC), a reduction of spine size and density in CA1 pyramidal neurons, an alteration in basic glutamatergic synaptic transmission and enhanced long-term depression (LTD) in the same neurons. In two lines of transgenic models for tauopathies, tauP301L single transgenic and APP/PS1/tauP301L triple transgenic mice, caspase-3 generally could not be detected in neurons containing truncated tau [43] [47]. A small exception was revealed in a few glia-like cells which were positive for both truncated tau and caspase-3 in postnatal mice. There was a dramatic decrease in caspase-3 level at the early development stage [30]. The low level of caspase-3 expression continued to take place during adult stages in both wild type and transgenic mice. Moreover, it has been showed that the decrease of caspase-3 during development took place at the transcriptional level, since its mRNA level correlated with the level of caspase-3. The tauP301L single transgenic and APP/PS1/tauP301L triple transgenic model disclosed that caspase-3 does not take part in the truncation of tau at D421, indicating that there is a different, caspase-3-independent path of tau modifications [47]. Caspase-3 was only responsible for the generation of Aβ-42 but not for tau modifications. 4. Activated Caspase-3 and Alzheimer’s Disease Therapy It is known that apoptosis with the participation of caspases e.g. caspase-3 may be involved in pathogenesis of AD [48] [49]. The study of Pellegrini et al. [50] showed that APP may be a specific substrate for caspase-3 and caspase-6 which cleave the intracellular part of this peptide between Asp664 and Ala665 and lead to increase of Aβ level, a toxic protein which in AD is associated with apoptotic neuronal death. Current drugs used in the treatment of AD, such as antagonists of the NMDA receptor, like memantine, and inhibitors of acetylcholinesterase, donepezil and ladostigil are likely to modify the process of apoptosis involving caspases (ceaspase-3) level and activity (Figure 1) [51]-[68]. Memantine (3,5-dimethyladamantan-1-amine) is the first in a novel class of medications acting by blocking NMDA-type glutamate receptors, introduced onto the German market in 1982. Memantine is approved by the U.S. FDA and the European Medicines Agency for AD treatment. It is hypothesized that glutaminergic dysfunction plays a crucial role in AD pathomechanism. It has been shown that excessive stimulation of glutamate N-methyl-D-aspartate (NMDA) receptors cause death of nerve cells due to excitotoxicity [51]. Memantine is a low and moderate-affinity noncompetitive antagonist of glutaminergic NMDA receptors. By binding to the NMDA receptor memantine is able to inhibit the prolonged influx of Ca2+ ions, which normalizes its function and protects the cell from activation of caspase-3 and apoptosis [52] [53]. The first clinical trials with memantine were carried out on a group of patients with moderate-to-severe AD [55]. During the one 24-week randomized analysis of patients with moderate AD, memantine occurred to be significantly more effective than A. Płóciennik et al. 67 Figure 1. Drugs and substances which may change caspase-3 expression in Alzheimer’s disease. placebo [56]. Because studies conducted so far have yielded conflicting results, memantine cannot be unequivocally labeled as useful in patients with moderate AD nor has it been officially registered for application to those patients [57]-[59]. Additional analyses post hoc regarding all previous parallel randomized controlled trials (RCTs), reveal a beneficial effect on improving cognition, functional ability and behavioral symptoms [60]-[66]. Donepezil, 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl] methylpiperidine hydrochloride, is reversible inhibitor of acetylcholinesterase which results in an increased level of acetylcholine in synapses. Donepezil produces improvements in reception and perception, colligation and deduction. The study carried out on single cells dissociated from the cerebral cortex of fetal rats showed its neuroprotective effect [67] [68]. It has been also shown that donepezil may decrease glutamate toxicity through down-regulation of NMDA receptors and Ca2+ influx induced by excessive glutamate, and subsequent activation of caspase-3 and neuronal death [67]. Ladostigil (TV3326) [(N-propargyl-(3R) aminoindan-5yl)-ethyl methyl carbamate] is a bifunctional drug having neuroprotecive, antiparkinsonian effect as both selective monoamine oxidase-(MAO)-B inhibitor and a cholinesterase inhibitor, which could be potentially administrated in AD therapy. The study of the potential antiapoptotic activity of ladostigil and its S-isomer TV3279 on an apoptotic model of long-term SK-N-SH neuroblastoma culture a dose-dependent showed decrease in viability due to prevention of caspase-3 activation [69]. However, treatment of neuroblastic cells with ladostigil lead to concentration-dependent decrease in intracellular APP levels compared with control untreated cells. Moreover, ladostigil did not affect APP mRNA levels, indicating posttranscriptional regulatory mechanism. Various inhibitors, including selective small molecules, selective peptidomimetic, selective non-peptide inhibitors of caspase-3 seem to be a potential drugs in therapy of AD (Figure 1) [70]-[79]. BZYX (acetylcholinesterase inhibitor), caspase-3, β-secretase, and γ-secretase inhibitors are now being preclinically tested in this disease. It is hopeful that BZYX(E-2-{4-[(Dimethylamino)methyl]benzylidene}-5,6-dimethoxy-2,3-dihydroinden-1-one) A. Płóciennik et al. 68 becomes a potential candidate for use in the intervention for neurodegenerative diseases. BZYX presented great neuroprotecive function from apoptosis, scavenged reactive oxygen species formation and resulted in up-regulation of procaspase-3 and down-regulation of phosphorylated JNK and p53 protein level and cleavage of caspase-3 [70]. Other substances with a potentially neuroprotecive effect of reducing caspases activity, such as caspase-3, have been designed several non-peptide inhibitors as well as small-molecule inhibitors, N-nitrosoaniline performing S-nitrosylation of the enzyme [78] [79]. In particular, isatin sulfonamides inhibitors were shown to inhibit selectively caspase-3 and -7. Other compounds, such as aza-peptide and sulfonamide isatin described as potential, irreversible and selective caspase inhibitors. From of these, only a few have been preclinically tested on animal models of human AD; their greatest drawback was their relatively limited selectivity. As of today, only VX-740 (Pralnacasan), VX-765, IDN-6556 and LB-84451 have been used in clinical tests [51] [52]. By the studies of early-onset AD, mutations in the PS gene were found, which suggests its responsibility for the development of AD [80]. PS1/2 is a transmembrane protein with enzymatic activity which forms γ-secretase with nicastrin complex. PS1/2 regulates intracellular Ca2+ levels, transport of proteins, β-catenin stability and protein degradation. Thus, various PS1/2 gene mutations have been suggested to be involved in Aβ42 generation, dysfunction of intracellular Ca2+ homeostasis and activation of apoptotic factors [80] [81]. The studies performed on human neuroblastoma cell line SH-SY5Y showed activation of apoptotic process, elevation of caspase-3 and -7 activities in mutant PSEN1-transfected cells. In addition the treatment with β- secretase or γ-secretase inhibitors, significantly alter the effects of the PSEN1 mutations on caspase-3 and -7 activations and improve cells viability [82]-[92]. Another potentional therapeutical strategy in AD is gene therapy [93] [94]. Studies on the protein seladin 1 can be an example [85]-[87]. Seladin 1 (product of DHCR24 gene located on 1p31.1-p33 chromosome) is a multifunctional enzyme with antioxidizing and antiapoptotic activity and, therefore, constitutes a major neuroprotective factor [95]. Seladin 1 is found in the endoplasmatic reticulum and, to a lesser degree, in the Golgie apparatus. Moreover, at the N-terminal segment of the protein an area was identified that may be indicative of its mitochondrial localization (MTS); there is no evidence, however, as to seladin 1 being present in mitochondria. This protein was identified with regard to its reduced expression in the cells affected by AD. Foreseeable, there was a positive correlation between seladin 1 overexpression and an increase in Aβ toxicity resistance during studies of cell cultures of H4 neuroglioma and embryonal SH-SY5Y neuroma [96] [97]. The study of Greeve et al. [96] on AD brains showed that seladin 1 overexpression protects cells from oxidizing stress and apoptosis caused by hydrogen peroxide and Aβ resulting in inhibition of caspase-3 activation. It was also found that under acute oxidative stress there is seladin 1 cleavage to 40 kDa protein by caspases (mainly caspase-6). In all probability the 40 kDa protein is inactive. Greeve et al. [96] put forward a hypothesis that both the 40 kDa product of seladin 1 cleavage and decrease in the DHCR24 gene transcription may induce a proapoptotic signal. 5. The Role of Caspase-3 in Synaptic Plasticity The role of caspase-3 in the nervous system exceeds the apoptosis. It was discussed, that except cell death this protein may be involved in several other processes, such as synaptic plasticity [98]. In 1998 caspase-3 was shown present in synaptic terminals [99]. Firstly it was supposed, that synaptic localization of caspase-3 is associated with apoptotic processes, spreading from terminals to cell body [100]. Further studies proposed that activation of caspases does not obligatory lead to apoptosis. The study of Kudryashova et al. [101] on Wistar rats’ hippocampal slices showed that in learning experiment, the caspase-3 activity reflects synaptic plasticity and not necessarily the apoptosis. Synaptic plasticity is a crucial process of modifying synaptic strength in response to neural activity and experience. Long-term potentiation (LTP) and long-term depression (LTD) are forms of synaptic plasticity, characterized by rapid changes in the synaptic strength of individual junctions [102]. The study of Gilman and Mattson [103] on neurons in apoptotic state have shown that activation of caspase-3 is followed by degradation of AMPA receptor, a part of R1 and R4 subunits of glutamate receptors, but not the NMDA receptors. Furthermore, it was shown that cultured hippocampal neurons treated with caspase-3 inhibitor exhibited increased function of AMPA receptors, what suggests that a low level of caspase activity is essential for reducing glutamate sensitivity in physiological conditions [104]. A study of Li et al. [105] merging overexpression of endogenous inhibitors and pharmacological inhibition of caspase-3 has shown that mitochondria mediated activation of caspase-3 is required for LTD in cell culture of CA1 hippocampal neurons. Subsequently, A. Płóciennik et al. 69 in hippocampal slices from caspase-3 knockout mice the LTD was abolished, whereas the LTP process seemed to be unaffected. Moreover, stimulation of NMDA receptor inducing LTD was followed by caspase-3 activation in dendrites, however without inducing further apoptosis. These data confirm a surprising pivotal role of caspase-3 in apoptosis and LTD. Furthermore, it was demonstrated in different animal models that caspase-3 also modifies learning ability and memory processes. Stephanichev et al. [106] used a caspase-3 inhibitor—ZDEVDFMK and showed that decreased level of caspase-3 in the adult rat brain impairs the acoustic startle response, providing strong evidence for a role of caspase-3 in normal cognitive processes in the mature brain. Huesmann and Clayton [107] have demonstrated that in birds active caspase-3 is vital for the forming of long-term habituation to a song. Subsequently, the inhibition of caspase-3 deteriorates the ability of song memorizing. Similarly, inhibition of caspase activity in the hippocampus has also been shown to impede long-term, but not short-term, spatial memory in the water maze task [108]. These reports are consistent with the emerging hypothesis that caspase-3 activation is required for learning and memory processes [103]. 6. The MicroRNA Dependent Regulation of Caspase-3 Activity MicroRNAs (miRNAs) are short RNA molecules consisting of 22 nucleotides, possessing regulatory functions [109]. They are responsible for fine-tuning of expression of various genes [110]. The mechanism of action is governed by binding of 6 nucleotide long “seed sequence” to particular, complementary fragments of an untranslated region (UTR) in 3’ end of mRNA strand with an assistance of miRNA induced silencing complex (miRISC). This reduces quantity of protein product [111]. Such mechanism was shown to be significant for adjusting the expression of approximately 60% of all human genes, e.g. encoding caspase-3 [112]. According to Tsang et al. [113] one of miRNA shown to be associated with caspase-3 activity was let-7a, a founding member in the let-7 family. It was demonstrated that ectopic expression of let-7a decreased the luciferase activity of a reporter construct containing the 3’-UTR of caspase-3 what was followed by repressed expression of the protein product in various human cancer cell lines: squamous carcinoma A431 and hepatocellular carcinoma HepG2. Moreover, in a doxorubicin-resistant A431 cell line (A10A subline) the down-regulation of caspase-3 was accompanied by let-7a overexpression. Furthermore, it was shown, that in lines expressing caspase-3 addition of anti-let-7a augmented proapototic effects of chemotherapeutic agents. Therefore, it is suspected that in cell cultures let-7a may control apoptosis by targeting caspase-3. Let-7c, the other member of let-7 family was shown by Ni et al. [114] to regulate caspase-3 activity in brain. They have used a mouse model of cerebral ischemia to find changes in expression of several inflammatory and proapoptotic proteins, including caspase-3. The researchers found a decreased expression of let-7c in plasma of investigated animals and ischemic patients, as well as in activated in vitro microglia. Subsequently, let-7c mimic was used to show that miRNA directly bind to the 3’-UTR of the caspase-3 mRNA, reducing levels of protein product. The caspase-3 plays a significant part in activation of microglia [115]. What is worth noting, microglia may play an important role in AD associated neuroinflamation [116]. Thus the down-regulation of let-7c may lead to neurodegeneration via caspase-3 and microglial activation. Peng et al. [117] have shown that in PC12 cells undergoing apoptosis following anoxia/reoxygenation (A/R) injury the expression of another let-7 family member, namely let-7e was decreased, what was accompanied by elevated expression of caspase-3. The scientists used the miRanda algorithm to identify putative binding sites of let-7e in the 3’-UTR of caspase-3. Subsequently in the PC12 cells transfected with pre-miRNA or let-7e the caspase-3 expression levels and cellular apoptosis following A/R injury were decreased, while co-transfection of anti-let-7e significantly decreased the effects of let-7e. It seems that let-7e inversely regulate caspase-3 expression and may be involved in A/R injury dependent apoptosis. Li T. et al. [118] have found another miRNA affected by A/R injury. In A/R treated H9C2 cardiomyocytes the expression of miR-30b was significantly decreased what was followed by increased caspase-3 level. The scientists confirmed a putative association by administration of mimic and inhibitor of miR-30b to cell culture. It was demonstrated that miR-30b mimic significantly decreased caspase-3 expression, whereas anti-miR-30b induced overexpression of the enzyme. It was also shown that miR-30b may protect the cardiomyocytes from apoptosis induced by A/R injury by inhibition of caspase-3 activity. The other member of miR-30 family, namely miR-30d was shown to control caspase-3 activity. Li N. et al. [119] have used bioluminescent caspase assay and they found that transfection with miR-30d mimic significantly reduced caspase-3 activity in cancer cells. It was also demonstrated that in transfected cells both total and A. Płóciennik et al. 70 Figure 2. Pathogenesis of Alzheimer’s disease and caspase-3 expression. cleaved caspase-3 protein was significantly reduced in both control and chemotherapeutic agent treated apoptotic cells. This suggests that miR-30d may be another miRNA controlling apoptosis in caspase-3 dependent manner. 7. Conclusions The molecular mechanism of neuronal damage in AD is not fully understood. It is known that in AD, with the progress of this disease there is a change in the level and expression of caspase-3 in various brain areas. Changes of caspase-3 expression may influence on impairment of many functions e.g. memory, learning, cognitive function of AD patients (Figure 2). Thus, it appears that caspase-3 may be used as diagnostic and prognostic factor in AD. It also seems that capsase-3 may be a potential target for pharmacological and gene therapies. Its level is modified by the pharmaceutical preparations currently used for the treatment of AD patients, including memantine, donepezil or ladostigil. However, now used medications in the treatment of AD are often ineffective. The understanding of the molecular mechanism of apoptosis involving caspase-3 participation seems to be important for finding effective AD therapies. Acknowledgements This work was supported by grants from Poznań University of Medical Sciences no. 502-05-01111677-50744 and 502-14-01111677-10342.
Activated Caspase-3 and Neurodegeneration and Synaptic Plasticity in Alzheimer’s Disease Anna Płóciennik, Michał Prendecki, Ewelina Zuba, Marcin Siudzinski, Jolanta Dorszewska Laboratory of Neurobiology, Department of Neurology, Poznan University of Medical Sciences, Poznań, Poland Email: dorszewskaj@yahoo.com Received 19 June 2015; accepted 25 August 2015; published 28 August 2015 Copyright © 2015 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract In Alzheimer’s disease (AD) there is a loss of neurons in different areas of the brain, including hippocampus, temporal and frontal cortex. Molecular mechanism of neuronal death in AD is not clear. It seems that apoptosis is a major form of neuronal cell death in this neurodegenerative disease. Neurons in both experimental animals and AD brains showed apoptotic changes, including DNA fragmentation, microRNA expression and caspase-3 activation. Activated caspase-3 may lead to amyloid precursor protein (APP) cleavage, extracellular senile plaques (SPs) and intracellular neurofibrillary tangles (NFTs) formation and neuronal loss, and it contributes to synaptic plasticity and cognitive dysfunction, especially in early stage of AD. Moreover, changes of caspase-3 activity were observed after therapy with memantine (NMDA receptor antagonist), donepezil and ladostigil (acetylcholinesterase inhibitors) in AD patients. It has been shown that both mementine and acetylcholinesterase inhibitors protect neurons from the caspase-3 activation. It seems that caspase-3 may be a potential diagnostic factor and target for pharmacotherapy of AD patients. Keywords Caspase-3, MicroRNA, Apoptosis, Synaptic Plasticity, Experimental Models, AD 1. Introduction Alzheimer’s disease (AD) is one of the most common neurodegenerative disorders of human beings. The disease manifests usually after the age of 65 years. The frequency of AD in this age group is 1% - 3% and doubles every five years, exceeding 40% in 85-year-old group. It is estimated that the number of patients suffering from A. Płóciennik et al. 64 AD is 21 - 24 million and this number will double in next 15 years. The disease occurs approximately two times more often in female than male [1]. AD usually lasts approximately for 10 years (less in men more for females). During the first 10 - 20 years the disease may be asymptomatic or manifest as mild cognitive impairment [2]. Then there is a growing mental process of memory and cognitive function may also experience changes in personality, delusions, confusion and depression that impair and ultimately prevent the independent functioning of the patient. Patients in last stage of the disease gradually lose the ability of communication, navigation, swallowing, etc. Patients become completely dependent on relatives and caregivers and require a constant supervision. The disease is usually fatal because of extracerebral complications [3]. There are two forms of AD, sporadic (SAD) with non-Mendelian genetic pattern and autosomaly dominantly inherited familial AD (FAD). Mutations of the amyloid precursor protein (APP), the presenilin 1 (PS1) and the presenilin 2 (PS2) coding genes (APP, PSEN1 and PSEN2, respectively) have been linked to FAD. Approximately 95% of AD cases occur as a SAD form, and the rest have autosomal dominant inheritance (FAD) [4] [5]. SAD was shown to be a multifactorial disease, in 70% determined by genetic factors and in 30% determined by non-genetic factors, e.g. environmental factors [6]. The most prominent genetic factors may be accounted genes involved in cholesterol metabolism and tricking, e.g. ABCA1, ABCA7, APOE and SORL1 [7] [8]. Of these genes APOE seems to be the most significant, where APOE ε4 allele is responsible for approximately 1/4 of SAD cases. It was also demonstrated that APOE ε4 may augment formation of senile plaques and impair clearance of AD pathological proteins [9]. The AD has been also associated with rare variants in other genes e.g. CR1, EPHA1, HLA, INPP5D, MS4A, TREM2 and TREM2L, responsible for immunology processes [10] and TP53-regulating apoptosis [11]. The most recent data indicate that AD may be also associated with dysregulation in microRNA expression [7] [12]. The most important non-genetic risk factors of AD include: being female, age over 65 years, low level of education, a poor nutritional status, a presence of relatives suffering from AD, an aluminum intoxication, chronic use of benzodiazepines, a tobacco use, a head trauma with loss of consciousness, a brain ischemia and infection of the central nervous system [13]. The most vulnerable areas in AD brains are the hippocampus and temporal, frontal, entorhinal cortex, and amygdale [14]. Neuropathological hallmarks of AD are neuronal loss, extracellular senile plaques (SPs), containing β-amyloid (Aβ) and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau proteins. It is known that Aβ accumulated in SPs is neurotoxic [15]. The Aβ is produced in brain in the manner of proteolysis. In physiological conditions—90% of APP, a transmembrane neuronal protein is cleaved by two enzymes: α- and γ-secretases, whose active subunits are coded by ADAM10, PSEN1 and PSEN2 genes, respectively. This leads to production of non-toxic proteolysis products [16]. In AD the second mechanism of APP lysis by β- and γ-secretases is activated, leading to formation of 42 aminoacid, highly hydrophobic Aβ. This peptide undergoes quick oligomerization and if the clearance mechanisms are disturbed (e.g. due to rare variants in respective genes), the oligomers bind to form larger structures—neurotoxic SPs [17]. Hyperphosphorylation of tau protein results in paired helical filaments (PHFs) formation which agglomerates in pericarions and creates NFTs which lead to disruption of intracellular transport [18]. Furthermore, NFTsbearing neurons have been suggested to be responsible for neuronal death in AD [19]. Data in literature indicated that Aβ and NFTs induce oxidative stress in neurons, and thus may be associated with neuronal loss in AD [18] [19]. Another neuropathological lesion in AD is granulovacuolar degeneration [20]. Up till now, the cause of damage to neurons and memory loss in the pathogenesis of AD is not clearly explained. One possible cause of neuronal loss in AD appears to be increased process of apoptosis [21] [22]. 2. The Role of Activated Caspase-3 in Pathogenesis of Alzheimer’s Disease Several studies [21]-[23] showed that apoptosis leads to neuronal death in AD. Changed expression of proteins linked to apoptosis, such as Bad, Bcl-2, Bax, p53 and Fas was reported in AD brains [24] [25]. Caspase-3, a protease, is thought to be an essential executioner caspase in the apoptosis [26]. Forming of active caspase-3 performs an irreversible step of the apoptotic pathway. Numerous studies have presented elevated level of caspase-3 in AD [27]-[30]. Su et al. [27] showed elevated active caspase-3 expression within SPs in postmortem hippocampus, frontal and entorhinal cortex of AD brains. Another studies showed that caspase-3 is prominent caspase implicated in A. Płóciennik et al. 65 APP cleavage [31]. Moreover, Gervais et al. [31] indicated that activated caspase-3 may lead to SPs formation, as well as it may play an essential role in neuronal loss in AD. The study of Khan et al. [4] showed elevated level of Aβ associated with increased caspase-3 activation in cytoplasmic hybrid cells created from mitochondrial DNA of SAD patients. Moreover, in this study secretion of Aβ was lowered by caspase-3 inhibition by incubation it with an antioxidant, pramipexol. According to the authors, mitochondrial genes in SAD may be responsible for Aβ oversecretion mediated by casapse-3 [4]. However, the study of Gastard et al. [28] indicated an increased expression of caspase-3 immunoreactivity in postmortem hippocampus, entorhinal cortex and cornu amonis of AD brains. In this study expression of activated caspase-3 was significantly higher in early stage of AD and it was colocalised with PHF-bearing neurons. Moreover, activated caspase-3 was lowered in advanced AD and in late stage of this disease was colocalised with classic NFTs. Su et al. [27] also demonstrated elevated levels of activated casapse-3 in postmortem hippocampus, frontal and etorhinal cortex and a prominent colocalisation between active casapse-3 and NFT-bearing neurons (pre-tangle neurons, early stage NFTs and mature NFTs). Another study showed that caspase-3 may be implicated in tau protein phosphorylation as well [32]. It seems that caspase-3 may be mediated NFTs formation and neuronal death in AD. Caspase-3 immunoreactivity was also reported in granules of granulovacuolar degeneration (GVD) in AD [27]-[29]. Granulovacuoles are considered as autophagic in origin [33] [34]. Activated caspase-3 in GVD may indicate the presence of self-repair processes in neurons. An existence of active caspase-3 in GVD might be elucidated by a lysosomal activation of caspases [35] [36], involving an activation of apoptosis which may lead to cell death and neuronal loss in AD. This notion is supported by evidence that apoptosis and autophagy are related processes and that there are autophagic forms of apoptotic pathway [37] [38]. Kovacs et al. [30] showed elevated caspase-3 level on the human H4 neuroglioma cell line, expressing mutation in PSEN1 gene. Increased level of caspase-3 was a result of staurosporine (STS) treatment, which has a proapoptotic effect. Authors showed that caspase-3 activation leads to cleaving of PS1. In the study of Tesco et al. [39] also showed that caspase may induce cleavage of PS1 and interrupts the interaction of PS1 with β-catenin which may contribute to cell death. 3. Activated Caspase-3 in the Experimental Models of Alzheimer’s Disease Currently pathogenesis of AD can be more clearly explained using experimental models of AD. Transgenic mouse models are very often used in the study of many neurodegenerative diseases, such as AD, Parkinson’s or Huntington’s diseases. The animal models carry mutations in specific genes which are involved in the pathogenesis of neurodegenerative diseases [40]-[46]. It is known that casapse-3 plays a significant role in the apoptosis and its level is markedly increased in diseases where such phenomenon occurs e.g. AD. The elevated level of casapse-3 was observed in hPS2m-Tg mice (carrying the human mutant PS2) and hPS2w-Tg mice (carrying the human wild PS2) compare to non-Tg mice [42]. Also, the increase in casapse-3 level was observed in a Tg2576 transgenic mouse model, which harbors the human APPswe mutant allele linked to FAD [45]. In this mouse model a significant increase in caspase-3 activity was detected in Tg2576 mice at the age 3 months (young adult), but not at 2 months. In another mouse model of AD, transgenic PS/APP, which expressed both the Swedish double mutations of APP (K670N/M671L) and PS1 (PS1M146L), a markedly elevated level of caspase-3 was observed as well [46]. In AD one of the major clinical features is memory impairment. The hippocampus plays a crucial role in memory processes, thus an increased caspase-3 activity should be detected in this particular structure. Indeed, in each quoted transgenic mouse model an enhanced caspase-3 level was disclosed in the hippocampus. In hPS2m-Tg and hPS2w-Tg mice it was observed in hippocampus-dentate gyrus but also in the cerebral cortex [42]. In Tg2576 transgenic mouse model an increase of caspase-3 was detected in CA1 pyramidal neurons [45]. In PS/APP transgenic model [APP (K670N/M671L) and PSEN1 (PS1M146L)] the elevation of caspase-3 activity was disclosed not only in the hippocampus (especially in the stratum lucidum of the hippocampal CA3 area) but also in other brain regions, including cortex, striatum, thalamus, and brainstem [46]. Also at the ultrastructural level there have been reported morphological changes typical for the apoptosis. Indeed, adult PS/APP mice exhibited pyknotic caspase-3-positive neurons which revealed such apoptotic changes as DNA fragmentation, caspase-3 activation, caspase-cleaved spectrin generation, chromatin margination and condensation, chromatin balls, and nuclear membrane fragmentation. What is more, the majority of caspase-3- A. Płóciennik et al. 66 immunoreactive neurons were grossly shrunken or fragmented without recognizable processes. Notably, activated caspase-3 and caspase-3-cleaved spectrin were abundant in autophagic vacuoles, accumulating in dystrophic neuritis of PS/APP mice [46]. Because synapses are necessary in the transmission of neuronal impulses which are involved in memory processes, it is supposed that these neuronal structures should be particularly affected in AD, thus presenting a high level of casapse-3 activation. In Tg2576 mice caspase-3 was more abundant in the synaptosomal fraction than in total homogenate [45]. Caspase-3 immunoreactivity was found in dendritic spines and it was more abundant in Tg2576 neurons than in their wild-type counterparts. In the spine, the immunoprecipitates were specifically concentrated in the postsynaptic sites. No immunoreactivity however was disclosed in cell soma, nuclei, presynaptic terminals or surrounding glial projections in Tg2576 mice. The study of D’Amelio et al. [45] showed that active caspase-3 is involved in the removal of the GluR1 subunit of AMPA-type receptor from postsynaptic sites which may lead to alterations of glutamatergic synaptic transmission and plasticity and correlated with spine degeneration and a deficit in hippocampal-dependent memory. The study revealed that these molecular modifications of glutamatergic synaptic transmission triggered by an active caspase-3 were performed by the activation of calcineurin A. In vivo, an elevation of caspase-3 activity in the hippocampus of the 3-month-old Tg2576 was associated with an increase of both activity and processing of calcineurin when compared with the wild type mice. Calcineurin A is proteolytically activated by caspase-3 and is responsible for Aβ-mediated synaptic removal of AMPAR which leads to the loss of dendritic spines [45]. In Tg2576 mice the caspase-3 activity increase was matched by a deficit in hippocampal-dependent contextual fear conditioning (CFC), a reduction of spine size and density in CA1 pyramidal neurons, an alteration in basic glutamatergic synaptic transmission and enhanced long-term depression (LTD) in the same neurons. In two lines of transgenic models for tauopathies, tauP301L single transgenic and APP/PS1/tauP301L triple transgenic mice, caspase-3 generally could not be detected in neurons containing truncated tau [43] [47]. A small exception was revealed in a few glia-like cells which were positive for both truncated tau and caspase-3 in postnatal mice. There was a dramatic decrease in caspase-3 level at the early development stage [30]. The low level of caspase-3 expression continued to take place during adult stages in both wild type and transgenic mice. Moreover, it has been showed that the decrease of caspase-3 during development took place at the transcriptional level, since its mRNA level correlated with the level of caspase-3. The tauP301L single transgenic and APP/PS1/tauP301L triple transgenic model disclosed that caspase-3 does not take part in the truncation of tau at D421, indicating that there is a different, caspase-3-independent path of tau modifications [47]. Caspase-3 was only responsible for the generation of Aβ-42 but not for tau modifications. 4. Activated Caspase-3 and Alzheimer’s Disease Therapy It is known that apoptosis with the participation of caspases e.g. caspase-3 may be involved in pathogenesis of AD [48] [49]. The study of Pellegrini et al. [50] showed that APP may be a specific substrate for caspase-3 and caspase-6 which cleave the intracellular part of this peptide between Asp664 and Ala665 and lead to increase of Aβ level, a toxic protein which in AD is associated with apoptotic neuronal death. Current drugs used in the treatment of AD, such as antagonists of the NMDA receptor, like memantine, and inhibitors of acetylcholinesterase, donepezil and ladostigil are likely to modify the process of apoptosis involving caspases (ceaspase-3) level and activity (Figure 1) [51]-[68]. Memantine (3,5-dimethyladamantan-1-amine) is the first in a novel class of medications acting by blocking NMDA-type glutamate receptors, introduced onto the German market in 1982. Memantine is approved by the U.S. FDA and the European Medicines Agency for AD treatment. It is hypothesized that glutaminergic dysfunction plays a crucial role in AD pathomechanism. It has been shown that excessive stimulation of glutamate N-methyl-D-aspartate (NMDA) receptors cause death of nerve cells due to excitotoxicity [51]. Memantine is a low and moderate-affinity noncompetitive antagonist of glutaminergic NMDA receptors. By binding to the NMDA receptor memantine is able to inhibit the prolonged influx of Ca2+ ions, which normalizes its function and protects the cell from activation of caspase-3 and apoptosis [52] [53]. The first clinical trials with memantine were carried out on a group of patients with moderate-to-severe AD [55]. During the one 24-week randomized analysis of patients with moderate AD, memantine occurred to be significantly more effective than A. Płóciennik et al. 67 Figure 1. Drugs and substances which may change caspase-3 expression in Alzheimer’s disease. placebo [56]. Because studies conducted so far have yielded conflicting results, memantine cannot be unequivocally labeled as useful in patients with moderate AD nor has it been officially registered for application to those patients [57]-[59]. Additional analyses post hoc regarding all previous parallel randomized controlled trials (RCTs), reveal a beneficial effect on improving cognition, functional ability and behavioral symptoms [60]-[66]. Donepezil, 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl] methylpiperidine hydrochloride, is reversible inhibitor of acetylcholinesterase which results in an increased level of acetylcholine in synapses. Donepezil produces improvements in reception and perception, colligation and deduction. The study carried out on single cells dissociated from the cerebral cortex of fetal rats showed its neuroprotective effect [67] [68]. It has been also shown that donepezil may decrease glutamate toxicity through down-regulation of NMDA receptors and Ca2+ influx induced by excessive glutamate, and subsequent activation of caspase-3 and neuronal death [67]. Ladostigil (TV3326) [(N-propargyl-(3R) aminoindan-5yl)-ethyl methyl carbamate] is a bifunctional drug having neuroprotecive, antiparkinsonian effect as both selective monoamine oxidase-(MAO)-B inhibitor and a cholinesterase inhibitor, which could be potentially administrated in AD therapy. The study of the potential antiapoptotic activity of ladostigil and its S-isomer TV3279 on an apoptotic model of long-term SK-N-SH neuroblastoma culture a dose-dependent showed decrease in viability due to prevention of caspase-3 activation [69]. However, treatment of neuroblastic cells with ladostigil lead to concentration-dependent decrease in intracellular APP levels compared with control untreated cells. Moreover, ladostigil did not affect APP mRNA levels, indicating posttranscriptional regulatory mechanism. Various inhibitors, including selective small molecules, selective peptidomimetic, selective non-peptide inhibitors of caspase-3 seem to be a potential drugs in therapy of AD (Figure 1) [70]-[79]. BZYX (acetylcholinesterase inhibitor), caspase-3, β-secretase, and γ-secretase inhibitors are now being preclinically tested in this disease. It is hopeful that BZYX(E-2-{4-[(Dimethylamino)methyl]benzylidene}-5,6-dimethoxy-2,3-dihydroinden-1-one) A. Płóciennik et al. 68 becomes a potential candidate for use in the intervention for neurodegenerative diseases. BZYX presented great neuroprotecive function from apoptosis, scavenged reactive oxygen species formation and resulted in up-regulation of procaspase-3 and down-regulation of phosphorylated JNK and p53 protein level and cleavage of caspase-3 [70]. Other substances with a potentially neuroprotecive effect of reducing caspases activity, such as caspase-3, have been designed several non-peptide inhibitors as well as small-molecule inhibitors, N-nitrosoaniline performing S-nitrosylation of the enzyme [78] [79]. In particular, isatin sulfonamides inhibitors were shown to inhibit selectively caspase-3 and -7. Other compounds, such as aza-peptide and sulfonamide isatin described as potential, irreversible and selective caspase inhibitors. From of these, only a few have been preclinically tested on animal models of human AD; their greatest drawback was their relatively limited selectivity. As of today, only VX-740 (Pralnacasan), VX-765, IDN-6556 and LB-84451 have been used in clinical tests [51] [52]. By the studies of early-onset AD, mutations in the PS gene were found, which suggests its responsibility for the development of AD [80]. PS1/2 is a transmembrane protein with enzymatic activity which forms γ-secretase with nicastrin complex. PS1/2 regulates intracellular Ca2+ levels, transport of proteins, β-catenin stability and protein degradation. Thus, various PS1/2 gene mutations have been suggested to be involved in Aβ42 generation, dysfunction of intracellular Ca2+ homeostasis and activation of apoptotic factors [80] [81]. The studies performed on human neuroblastoma cell line SH-SY5Y showed activation of apoptotic process, elevation of caspase-3 and -7 activities in mutant PSEN1-transfected cells. In addition the treatment with β- secretase or γ-secretase inhibitors, significantly alter the effects of the PSEN1 mutations on caspase-3 and -7 activations and improve cells viability [82]-[92]. Another potentional therapeutical strategy in AD is gene therapy [93] [94]. Studies on the protein seladin 1 can be an example [85]-[87]. Seladin 1 (product of DHCR24 gene located on 1p31.1-p33 chromosome) is a multifunctional enzyme with antioxidizing and antiapoptotic activity and, therefore, constitutes a major neuroprotective factor [95]. Seladin 1 is found in the endoplasmatic reticulum and, to a lesser degree, in the Golgie apparatus. Moreover, at the N-terminal segment of the protein an area was identified that may be indicative of its mitochondrial localization (MTS); there is no evidence, however, as to seladin 1 being present in mitochondria. This protein was identified with regard to its reduced expression in the cells affected by AD. Foreseeable, there was a positive correlation between seladin 1 overexpression and an increase in Aβ toxicity resistance during studies of cell cultures of H4 neuroglioma and embryonal SH-SY5Y neuroma [96] [97]. The study of Greeve et al. [96] on AD brains showed that seladin 1 overexpression protects cells from oxidizing stress and apoptosis caused by hydrogen peroxide and Aβ resulting in inhibition of caspase-3 activation. It was also found that under acute oxidative stress there is seladin 1 cleavage to 40 kDa protein by caspases (mainly caspase-6). In all probability the 40 kDa protein is inactive. Greeve et al. [96] put forward a hypothesis that both the 40 kDa product of seladin 1 cleavage and decrease in the DHCR24 gene transcription may induce a proapoptotic signal. 5. The Role of Caspase-3 in Synaptic Plasticity The role of caspase-3 in the nervous system exceeds the apoptosis. It was discussed, that except cell death this protein may be involved in several other processes, such as synaptic plasticity [98]. In 1998 caspase-3 was shown present in synaptic terminals [99]. Firstly it was supposed, that synaptic localization of caspase-3 is associated with apoptotic processes, spreading from terminals to cell body [100]. Further studies proposed that activation of caspases does not obligatory lead to apoptosis. The study of Kudryashova et al. [101] on Wistar rats’ hippocampal slices showed that in learning experiment, the caspase-3 activity reflects synaptic plasticity and not necessarily the apoptosis. Synaptic plasticity is a crucial process of modifying synaptic strength in response to neural activity and experience. Long-term potentiation (LTP) and long-term depression (LTD) are forms of synaptic plasticity, characterized by rapid changes in the synaptic strength of individual junctions [102]. The study of Gilman and Mattson [103] on neurons in apoptotic state have shown that activation of caspase-3 is followed by degradation of AMPA receptor, a part of R1 and R4 subunits of glutamate receptors, but not the NMDA receptors. Furthermore, it was shown that cultured hippocampal neurons treated with caspase-3 inhibitor exhibited increased function of AMPA receptors, what suggests that a low level of caspase activity is essential for reducing glutamate sensitivity in physiological conditions [104]. A study of Li et al. [105] merging overexpression of endogenous inhibitors and pharmacological inhibition of caspase-3 has shown that mitochondria mediated activation of caspase-3 is required for LTD in cell culture of CA1 hippocampal neurons. Subsequently, A. Płóciennik et al. 69 in hippocampal slices from caspase-3 knockout mice the LTD was abolished, whereas the LTP process seemed to be unaffected. Moreover, stimulation of NMDA receptor inducing LTD was followed by caspase-3 activation in dendrites, however without inducing further apoptosis. These data confirm a surprising pivotal role of caspase-3 in apoptosis and LTD. Furthermore, it was demonstrated in different animal models that caspase-3 also modifies learning ability and memory processes. Stephanichev et al. [106] used a caspase-3 inhibitor—ZDEVDFMK and showed that decreased level of caspase-3 in the adult rat brain impairs the acoustic startle response, providing strong evidence for a role of caspase-3 in normal cognitive processes in the mature brain. Huesmann and Clayton [107] have demonstrated that in birds active caspase-3 is vital for the forming of long-term habituation to a song. Subsequently, the inhibition of caspase-3 deteriorates the ability of song memorizing. Similarly, inhibition of caspase activity in the hippocampus has also been shown to impede long-term, but not short-term, spatial memory in the water maze task [108]. These reports are consistent with the emerging hypothesis that caspase-3 activation is required for learning and memory processes [103]. 6. The MicroRNA Dependent Regulation of Caspase-3 Activity MicroRNAs (miRNAs) are short RNA molecules consisting of 22 nucleotides, possessing regulatory functions [109]. They are responsible for fine-tuning of expression of various genes [110]. The mechanism of action is governed by binding of 6 nucleotide long “seed sequence” to particular, complementary fragments of an untranslated region (UTR) in 3’ end of mRNA strand with an assistance of miRNA induced silencing complex (miRISC). This reduces quantity of protein product [111]. Such mechanism was shown to be significant for adjusting the expression of approximately 60% of all human genes, e.g. encoding caspase-3 [112]. According to Tsang et al. [113] one of miRNA shown to be associated with caspase-3 activity was let-7a, a founding member in the let-7 family. It was demonstrated that ectopic expression of let-7a decreased the luciferase activity of a reporter construct containing the 3’-UTR of caspase-3 what was followed by repressed expression of the protein product in various human cancer cell lines: squamous carcinoma A431 and hepatocellular carcinoma HepG2. Moreover, in a doxorubicin-resistant A431 cell line (A10A subline) the down-regulation of caspase-3 was accompanied by let-7a overexpression. Furthermore, it was shown, that in lines expressing caspase-3 addition of anti-let-7a augmented proapototic effects of chemotherapeutic agents. Therefore, it is suspected that in cell cultures let-7a may control apoptosis by targeting caspase-3. Let-7c, the other member of let-7 family was shown by Ni et al. [114] to regulate caspase-3 activity in brain. They have used a mouse model of cerebral ischemia to find changes in expression of several inflammatory and proapoptotic proteins, including caspase-3. The researchers found a decreased expression of let-7c in plasma of investigated animals and ischemic patients, as well as in activated in vitro microglia. Subsequently, let-7c mimic was used to show that miRNA directly bind to the 3’-UTR of the caspase-3 mRNA, reducing levels of protein product. The caspase-3 plays a significant part in activation of microglia [115]. What is worth noting, microglia may play an important role in AD associated neuroinflamation [116]. Thus the down-regulation of let-7c may lead to neurodegeneration via caspase-3 and microglial activation. Peng et al. [117] have shown that in PC12 cells undergoing apoptosis following anoxia/reoxygenation (A/R) injury the expression of another let-7 family member, namely let-7e was decreased, what was accompanied by elevated expression of caspase-3. The scientists used the miRanda algorithm to identify putative binding sites of let-7e in the 3’-UTR of caspase-3. Subsequently in the PC12 cells transfected with pre-miRNA or let-7e the caspase-3 expression levels and cellular apoptosis following A/R injury were decreased, while co-transfection of anti-let-7e significantly decreased the effects of let-7e. It seems that let-7e inversely regulate caspase-3 expression and may be involved in A/R injury dependent apoptosis. Li T. et al. [118] have found another miRNA affected by A/R injury. In A/R treated H9C2 cardiomyocytes the expression of miR-30b was significantly decreased what was followed by increased caspase-3 level. The scientists confirmed a putative association by administration of mimic and inhibitor of miR-30b to cell culture. It was demonstrated that miR-30b mimic significantly decreased caspase-3 expression, whereas anti-miR-30b induced overexpression of the enzyme. It was also shown that miR-30b may protect the cardiomyocytes from apoptosis induced by A/R injury by inhibition of caspase-3 activity. The other member of miR-30 family, namely miR-30d was shown to control caspase-3 activity. Li N. et al. [119] have used bioluminescent caspase assay and they found that transfection with miR-30d mimic significantly reduced caspase-3 activity in cancer cells. It was also demonstrated that in transfected cells both total and A. Płóciennik et al. 70 Figure 2. Pathogenesis of Alzheimer’s disease and caspase-3 expression. cleaved caspase-3 protein was significantly reduced in both control and chemotherapeutic agent treated apoptotic cells. This suggests that miR-30d may be another miRNA controlling apoptosis in caspase-3 dependent manner. 7. Conclusions The molecular mechanism of neuronal damage in AD is not fully understood. It is known that in AD, with the progress of this disease there is a change in the level and expression of caspase-3 in various brain areas. Changes of caspase-3 expression may influence on impairment of many functions e.g. memory, learning, cognitive function of AD patients (Figure 2). Thus, it appears that caspase-3 may be used as diagnostic and prognostic factor in AD. It also seems that capsase-3 may be a potential target for pharmacological and gene therapies. Its level is modified by the pharmaceutical preparations currently used for the treatment of AD patients, including memantine, donepezil or ladostigil. However, now used medications in the treatment of AD are often ineffective. The understanding of the molecular mechanism of apoptosis involving caspase-3 participation seems to be important for finding effective AD therapies. Acknowledgements This work was supported by grants from Poznań University of Medical Sciences no. 502-05-01111677-50744 and 502-14-01111677-10342.







论文发表 是一个专门从事期刊推广、论文发表、论文发表辅导的机构。融合收集数百家期刊杂志社征稿评职称,发表论文,以供广大作者免费阅览,以期待各位作者在短时间内掌握每种期刊征稿要求、审稿范围,在日常繁重的工作中短时间开心、放心快速投稿、在适合的时间拿到刊物,评上职称,不为挤不上评职称的末班车而烦恼。
本站主要整合如下评职称,发表论文:
教育论文发表
经济论文发表
科技论文发表
医学论文发表
计算机论文发表
文学论文发表
农业论文发表
学报论文发表
其它论文发表等,另期待更多的杂志编辑与本站联系文:
85782530(普刊发表)
82534308(核心发表)共同以正期刊界正刊之气,维护学术良好的氛围。
免责声明:本网所提供的信息资源如有侵权、违规,请及时告知!
论文网版权所有, 本站提供论文发表 论文投稿 发表论文 发表文章
文章只代表作者观点,并不意味着本站认同,部分作品系转载,版权归原作者或相应的机构;若某篇作品侵犯您的权利,请来信告知:lunww@126.com
中国论文网全权所属国际域名:www.lunww.com 中文域名:论文发表 技术支持:论文发表 国家双软认定单位 法律顾问:全民安律师事务所
期刊合作加盟咨询:5715378(加盟、投诉、建议) 论文发表咨询电话:18262951856 论文发表投稿邮箱:lunww@126.com
Powered by 论文发表 © 2010-2020

苏ICP备19023845号-4