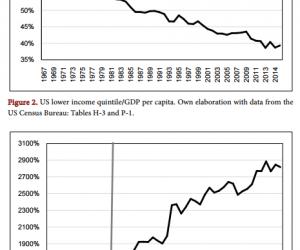
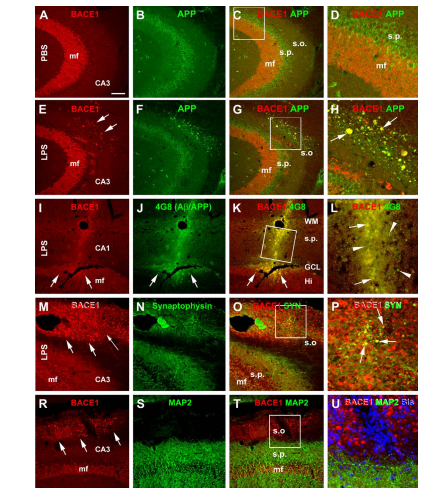
 Lipolysaccharide-Induced Neuroinflammation Is Associated with Alzheimer-Like Amyloidogenic Axonal Pathology and Dendritic Degeneration in Rats Xiaohua Deng1*, Meili Li1*, Weiming Ai1,2, Lixin He1,3, Dahua Lu1, Peter R. Patrylo4, Huaibin Cai5, Xuegang Luo1, Zhiyuan Li1, Xiao-Xin Yan1# 1 Department of Anatomy and Neurobiology, Central South University School of Basic Medical Science, Changsha, China 2 Department of Nursing in Internal Medicine, School of Nursing, Xiangtan Vocational and Technical College, Xiangtan, China 3 Department of Anatomy and Physiology, School of Nursing, Xiangtan Vocational and Technical College, Xiangtan, China 4 Departments of Physiology, Anatomy and the Center for Integrated Research in Cognitive and Neural Sciences, Southern Illinois University Carbondale, Carbondale, USA 5 Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, USA Email: # yanxiaoxin@csu.edu.cn Received 9 May 2014; revised 31 May 2014; accepted 7 June 2014 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Chronic neuroinflammation is thought to play an etiological role in Alzheimer’s disease (AD) which is characterized pathologically by amyloid and tau formation, as well as neuritic dystrophy and synaptic degeneration. The causal relationship between these pathological events is a topic of ongoing research and discussion. Recent data from transgenic AD models point to a tight spatiotemporal link between neuritic and amyloid pathology, with the obligatory enzyme for β-amyloid (Aβ) production, namely β-secretase-1 (BACE1), being overexpressed in axon terminals undergoing dystrophic change. However, the axonal pathology inherent with BACE1 elevation seen in transgenic AD mice may be secondary to increased soluble Aβ in these genetically modified animals. Further, it is unclear whether the inflammation seen in AD is the result of , or the cause of * Authors contributed to this work equally. # Corresponding author. X. H. Deng et al. 79 neuritic dystrophy. Here we explored the occurrence of AD-like axonal and dendritic pathology in adult rat brains affected by LPS-induced chronic neuroinflammation. Unilateral intracerebral LPS injection induced prominent inflammatory response in glial cells in the ipsilateral cortex and hippocampal formation. BACE1 protein levels were elevated in the ipsilateral hippocampal lysates in the LPS-treated animals relative to controls. BACE1 immunoreactive dystrophic axons appeared in the LPS-treated ipsilateral cortex and hippocampal formation, colocalizing with increased β-amyloid precursor protein and Aβ antibody (4G8) immunolabeling. Quantitative Golgi studies revealed reduction of dendritic branching points and spine density on cortical layer III and hippocampal CA3 pyramidal neurons in the LPS-treated ipsilateral cerebrum. These findings suggest that Alzheimer-like amyloidogenic axonal pathology and dendritic degeneration occur in wildtype mammalian brain in partnership with neuroinflammation following LPS injection. Keywords Amyloid Pathogenesis, Neuritic Dystrophy, Neurodegeneration, Neuroplasticity, Synaptic Pathology 1. Introduction Neuroinflammation has been linked to many neuropsychiatric disorders, including neurodegenerative diseases such as Alzheimer’s diseases (AD), Parkinson’s disease (PD), multiple sclerosis and traumatic brain injury [1]- [8]. Aging is a major risk factor for many age-related diseases, and is associated with a certain degree of chronic inflammation [9] [10]. In general, chronic inflammation is considered to mount lasting stress on neurons and synapses, and may lead to brain dysfunction, including cognitive deficits [5] [11]-[14]. The causal relationship between chronic inflammation and some of the hallmark pathological lesions in neurological diseases is under intensive investigation. For example, oxidative or inflammatory stress is suggested to promote cerebral amyloid pathology via increased production and/or impaired clearance of Aβ, involving both neurons and glial cells [15]-[19]. Aβ products including soluble and aggregated variants may also act as proinflammatory factors [20] [21]. Loss of synapses and their connectivity best correlate with cognitive deficits in AD [22]-[29]. The density of dendritic spines appears to be reduced readily at prodromal stages of the disease [30]. Besides synaptic degeneration, axonal elements including presynaptic terminals undergo aberrant sprouting and dystrophic expansion [30]-[32]. Recent data from transgenic AD models, nonhuman primates and human subjects show that upregulation of the amyloidogenic proteins, especially the rate-limiting enzyme β-secretase-1 (BACE1), appears to be an molecular cascade tightly associated with axonal sprouting and dystrophy, suggestive of a driving role for amyloidogenic axonal pathology in plaque formation [33]-[35]. The bacterial endotoxin lipopolysaccharide (LPS) can induce chronic neuroinflammation in rodents [36]-[38]. LPS administration also causes learning and memory deficits in the animals, providing an excellent model system for studying cognitive dysfunction associated with chronic neuroinflammation [39]-[41]. Neuroinflammation is considered to play an early or inductive role in the development of AD pathologies, although the anatomic evidence remains to be better formulated. Therefore, the present study was set to address whether intracerebral LPS injection in adult rats may induce axonal and dendritic pathologies similar to that seen in AD [32]. Specifically, we aimed to identify whether dystrophic axonal pathology inherent with amyloidogenic modulation and degenerative dendritic/spine changes occur on cerebral principal neurons in LPS-treated adult rats. 2. Materials and Methods 2.1. Animals and Intracerebral Injection In-house bred male adult Sprague-Dawley rats (n = 24) weighing ~200 g (Animal Center of Central South University) were used. Rats were maintained in temperature (20˚C - 26˚C), humidity (30% - 60%) and lighting (12:12 hours light/dark cycle) controlled rooms, with food and water freely available. For intracerebral injections, rats were placed in a stereotaxic apparatus under sodium pentobarbital anesthesia (50 mg/kg, i.p.). LPS X. H. Deng et al. 80 from Escherichia coli serotype 055:B5 (L2637, Sigma-Aldrich, St. Louis, MO, USA) was dissolved (2.5 µg/µl) in sterile phosphate-buffered saline (PBS, 0.01 M, pH 7.2) (vehicle) [41]. LPS (10 µg in 4 µl) was injected through the neocortex into the right hippocampus with a microsyringe in each animal (n = 12), using the following coordinates: 3.5 mm lateral to the sagittal cranial suture; 5 mm caudal to the bregma and 3 mm below the dura mater. Controls (n = 12) were injected with the same amount of PBS. Brain examination was carried out 30 days post intracortical injection. The experimental protocol was approved by the Animal Care and Use Committee of the Central South University and were in compliance with NIH guidelines. 2.2. Immunoblot Hippocampi ipsilateral to the injection side were dissected out following decapitation (n = 4/group). Tissue was weighed and homogenized in a commercial protein extraction buffer at 1 to 10 w/v ratio (CW0883, Kangwei Century Company, Beijing), followed by centrifuge at 15,000 g. Protein concentrations in the supernatants were determined by DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA). A total of 25 µg protein from each sample was separated electrophoretically in 10% SDS-polyacrylamide gel and transferred to polyvinylidene fluoride membrane. Membranes were blocked with 1% non-fat milk and 5% bovine serum albumin (BSA) in 0.1 M Tris-buffered saline containing 0.1% Tween-20 (TBS-T) for 2 hours at room temperature. Membranes were then incubated overnight at 4˚C in the same buffer containing rabbit anti-BACE1 (1:2000) [33] [34] [42] [43], rabbit anti-glial fibrillary acidic protein (GFAP) (G9269, Sigma-Aldrich, St Louis, MO, USA, 1:4000) or rabbit anti-β-tubulin (Sigma-Aldrich, T2200, 1:10000). Membranes were washed thoroughly with TBS-T, then were incubated for 2 hours at room temperature with horseradish peroxidase-conjugated goat anti-rabbit IgGs (1:20,000; Bio-Rad Laboratories). Bound antibodies were detected by enhanced chemiluminescence (ECL kit, GE Healthcare Life Sciences, Piscataway, NJ, USA). The membranes were exposed to X-ray films developed subsequently in a darkroom. The films were scanned, with optical density (OD) of immunoblot bands measured using Image-J, followed by standardization to the internal references. 2.3. Immunohistochemistry Rats were perfused transcardially with PBS followed by 4% phosphate-buffered (0.1M) paraformaldehyde (pH 7.4). Brains were removed and postfixed for 12 hours and transferred into 30% sucrose for cryoprotection. Thirty micrometer-thick coronal sections were cut in a cryostat, with 12 sets of sections collected for cresyl violet stain and for immunohistochemical studies. For immunohistochemistry with the avidin-biotin complex (ABC) method, free-floating sections were soaked in 1% H2O2 in PBS for 30 minutes to diminish endogenous peroxidase activity, and preincubated in PBS buffer containing 0.2% Triton X-100 and normal horse serum for 1 hour. The sections were then reacted with one of the following primary antibodies: 1) Mouse monoclonal antibody against major histocompatibility complex class II molecules (MHC-II) at 1:1000 (ab55152, Abcam, Cambridge, MA, USA); 2) Rabbit anti-human BACE1 at 1:2000; 3) mouse anti-β-amyloid precursor protein (APP) monoclonal antibody 22C11 (MAB348, EMD Millipore, Billerica, MA, USA, 1:4000); 4) mouse anti-Aβ17-24 monoclonal antibody 4G8 (#39240, Signet, Dedham, USA, 1:4000); 5) rabbit anti-GFAP (G9269, Sigma-Aldrich, 1:2000). The sections were further reacted with a biotinylated pan-specific secondary antibody (BA-1300, Vector Laboratories, Burlingame, CA, USA, 1:400) for 2 hours, and subsequently with the ABC reagents (PK-6100, Vector Laboratories, 1:400) for one hour. Immunoreactivity was visualized in 0.05% 3,3’-diaminobenzidine (DAB) and 0.003% H2O2. Selected sections were processed for double immunofluorescence beginning with blocking nonspecific reactivity by incubation in PBS buffer containing 5% normal donkey serum. Sections were further reacted overnight at 4˚C with a pair of primary antibodies raised in different species in PBS containing 0.2% Triton X-100 and the blocking serum. The antibody pairs included: 1) mouse anti-MCH-II (1:1000) and rabbit anti-CD11b (MABT149, EMD Millipore, 1:1000); 2) mouse anti-MCH-II and rabbit anti-GFAP (G9269, 1:2000); 3) rabbit anti-BACE1 and mouse anti-APP 22C11; 4) rabbit anti-BACE1 and mouse anti-Aβ 4G8; 5) rabbit anti-BACE1 and mouse anti-synaptophysin (MAB329, EMD Millipore, 1:4000); 6) rabbit anti-BACE1 and mouse anti-microtubule associated protein-2 (MAP2) (M9942, Sigma-Aldrich, 1:2000). On the second day, the sections were rinsed with PBS and incubated at room temperature for 2 hours with Alexa Fluor® 488 and Alexa Fluor® 594 conjugated donkey anti-mouse and anti-rabbit IgGs (1:200, Invitrogen, Carlsbad, CA, USA). Sections were then counter-stained with bisbenzimide (Hoechst 33342, 1:50000, Catalog #B2261, Sigma-Aldrich), washed thoroughly, and mounted with anti-fading medium before microscopic examination. X. H. Deng et al. 81 2.4. Rapid Golgi-Cox Stain Brains were removed following a vascular rinse with PBS. Blocks containing the middle 1/3 cerebrum of both hemispheres were rinsed briefly in double distilled water, and processed with the FD Rapid Golgi stain TM Kit (FD Neuro Technologies, Ellicott City, MD) following the manufacturer’s instruction. The brain blocks were immersed in freshly made mixture of Solutions A and B (1:1) in the dark at room temperature for 2 weeks, and then in Solution C at 4˚C in the dark for 3 days. After silver impregnation, the blocks were cut slowly into frontal sections at 100 μm thickness in a vibratome. Sections were collected alternatively in 10 sets in Solution C, mounted on gelatin-coated microslides, dehydrated through ascending concentrations of ethanol, cleared in xylene, and sealed with PermountTM mounting medium. 2.5. Imaging and Data Analysis An Olympus (BX53) microscope equipped with imaging system (CellSens Standard, Olympus) was used for examining sections stained with the ABC and fluorescent methods. Double immunofluorescence was also imaged on a confocal microscope (Nikon, DIGITAL ECLIPSE C1 plus, 5 μm thickness scan). Immunolabeling in sections around the level of injection in the rostrocaudal dimension was comparatively examined between the ipsilateral and contralateral cerebral hemispheres, using the needle track as a reference. Golgi-impregnated sections were examined on a Zeiss Axioplan microscope equipped with the Neurolucida and a high-resolution motorized stage for 3D neuronal reconstruction (MicroBrightField China). Two sections nearest to the injection coordinates (in the rostrocaudal dimension) were selected from each brain for neuronal morphometric analysis. Golgi-stained pyramidal neurons that met the following criteria were selected for reconstruction: 1) they were located in layer III of the parietal cortex overlying the mid-hippocampus and in the middle portion of CA3 (i.e., around the dorsal to ventral turning area); 2) they were among the labeled cells with the widest dendritic field by overall visual judgment; 3) they were well separated from other impregnated cells such that their dendritic tree was not or minimally overlapped with the processes from other cells; 4) the somata and dendritic processes were well-impregnated throughout the section thickness, with no apparent truncation of the dendritic arbor. Ten pyramidal neurons per region/brain were reconstructed with the aid of the Neurolucida software. Subsequently, somal area, total length of the dendritic tree, branching nodes and spine density (per 10 μm length) of the apical and basal dendrites, were obtained from a given selected neuron. 2.6. Statistical Analysis Imaging and numerical data for comparing groups were processed, with the mean ± SD calculated. Means were statistically analyzed by Student-t test or one-way ANOVA with posthoc Duncan’s multi-group comparisons when applicable. P < 0.05 was considered statistically significant. Figures were assembled with Photoshop 7.1, with brightness and contract adjusted as needed. 3. Results 3.1. LPS Injection Induced Immunoinflammatory Cellular/Molecular Changes To confirm the occurrence of chronic neuroinflammation, cerebral sections from the LPS and PBS groups were processed under identical conditions for the detection of immunoinflammatory proteins. Compared to PBS controls, increased immunoreactivity for MCH-II (Figures 1(A)-(E)), CD11b (not shown) and GFAP (not shown) occurred in the ipsilateral cortex and hippocampal formation in the LPS-injected brain sections. Specifically, MCH-II immunoreactive cells appeared to be largely glial cells (Figure 1(D) and Figure 1(E)). Double immunofluorescence showed that the majority of MCH-II labeled cells co-expressed the microglial marker CD11b (Figures 1(F)-(H)), although a few also colocalized with GFAP immunoreactivity, suggestive of a colocalization in astrocytes (Figures 1(I)-(K)). 3.2. LPS Injection Elevated BACE1 Protein Levels BACE1 protein levels were immunoblotted with a well-characterized rabbit antibody, which detects mature BACE1 protein migrating at ~70 kd [33] [34] [42] [43]. In the lysates of the ipsilateral hippocampi, immunob- X. H. Deng et al. 82 Figure 1. Representative images illustrating immunoinflammatory cellular/molecular changes in rat cortex and hippocampal formation following intracerebral lipolysaccharide (LPS) injection. Panel (A) shows minimal immunoreactivity of major histocompatibility complex class II molecules (MHC II) in the hippocampal CA1 to CA3 areas, the dentate gyrus (DG) and the overlaying cortex (Ctx) in a PBS-injected control rat, with the area of the dentate gyrus enlarged as (C). MHC II immunolabeling is increased in both the cortex and hippocampal formation in the LPS-injected animal (B), with the labeled cellular profiles appeared as glial cells at high magnifications ((D), (E)). Confocal double immunofluorescence shows a great extent of colocalization of MHC II reactivity among CD11b labeled microglial cells in the LPS-treated cerebrum ((F)-(H)). A small amount of MHC II labeled cells exhibit glial fibrillary acidic protein (GFAP) immunoreactivity, suggestive of a colocalization in activated astrocytes ((I)-(K)). Scale bar = 500 μm in A applying to B; equivalent to 250 μm for ((C), (D)); and to 75 μm for ((E)-(K)). lotted BACE1 signal was significantly increased relative to PBS-treated counterparts (Figure 2(A)). The mean optic density of BACE1 in the LPS group increased one fold (199.0% ± 52.8%) relative to control (100.0% ± 12.2%) (P = 0.043, t = 3.40, df = 3; two-tailed paired t-test) (Figure 2(A) and Figure 2(B)). Serving as an experimental as well as assay control [36]-[38], GFAP levels in the same cerebral samples were checked. In the ipsilateral hippocampal lysates, levels of GFAP were significantly (P = 0.027, t = 4.03, df = 3; two-tailed paired t-test) elevated in the LPS-treated (141.7% ± 11.4%) relative to PBS-treated (100% ± 13.4%) samples (Figure 2(A) and Figure 2(C)). 3.3. LPS Injection Induced Axonal Pathology with Enhanced Amyloidogenic Protein Expression In normal mammalian brains BACE1 immunoreactivity is largely expressed in the neuropil in a diffuse pattern X. H. Deng et al. 83 Figure 2. Western blot analysis showing elevation of the levels of β-secretase-1 (BACE1) and GFAP in the LPS-injected relative to PBS-treated (control) brains (n = 4/group), as assayed using hippocampal lysates ipsilateral to the injection. BACE1 levels in the LPS-treated group are about 2 times of that in the control ((A), (B)), while GFAP levels in the LPS group approach to 1.5 times of the control ((A), (C)), with the changes being significant for both proteins (paired two-tail t-tests). except for a distinct heavy labeling at selected neuronal terminal fields including the mossy fiber terminals and olfactory glomeruli. In human and transgenic animal brains with amyloid plaque pathology, BACE1 immunoreactivity is increased preferentially in swollen and sprouting axonal terminals or axonal dystrophic neurites [33]-[35] [42]. In the present study, BACE1 labeling in both cerebral hemispheres in the PBS injected brains and the contralateral cerebrum in the LPS-injected animals exhibited the aforementioned normal distribution pattern (Figure 3(A) and Figure 3(B)). On the contrary, BACE1 labeled neuritic structures emerged in the ipsilateral hemisphere of the LPS-treated brains in a site-specific manner. Thus, increased BACE labeling appeared in both the cortex and hippocampal formation especially evident around the needle track. At high magnification, these labeled profiles appeared as swollen sphericles and neuritic processes (Figures 3(B)-(F)). In the white matter, these neuritic processes appeared to spread from the needle track for a considerable distance (Figure 3(B) and Figure 3(D)). In the ipsilateral hippocampus, the BACE1 labeled swollen neurites occurred in the stratum pyramidale (s.p.) to stratum-lacunosum-moleculare (s.l.m.) of CA3 and the adjacent CA1 area (Figure 3(C)). A large amount of swollen sphericles extended along the stratum oriens (s.o.) of CA3 in the LPS-injected animals (Figure 3(E) and Figure 3(F)). At high magnifications, these BACE1 labeled neurites were mostly round or oval in shape, and varied in size. Some smaller sphericles were sometimes arranged in chains resembling enlarged axonal varicosities (Figure 3(C), Figure 3(D) and Figure 3(F)). In double immunofluorescence, the BACE1 immunoreactive swollen neurites exhibited a great extent of colocalization with APP (Figures 4(A)-(H)). In addition, the Aβ antibody 4G8 (which detects mouse APP, Aβ and likely APP β-C-terminal fragments, ref. 34) showed increased immunoreactivity in the same area with BACE1 labeled neurites (Figures 4(I)-(K)). A colocalization of BACE1 and 4G8 labeling was detected among some individual neuritic profiles. Notably, there existed a considerable amount of diffuse extracellular Aβ labeling (not colocalized with BACE1 reactivity), some of which appeared punctate (Figure 4(L)). The BACE1 labeled X. H. Deng et al. 84 Figure 3. Representative images illustrating the occurrence of BACE1 immunoreactive neuritic pathology after focal LPS injection in rat cerebrum. In the vehicle control (injection of phosphate-buffered saline, PBS), the pattern of BACE1 immunoreactivity is comparable between the ipsilateral and contralateral cortex and hippocampal formation, which is generally associated with neuropil except for a heavy labeling at the hippocampal mossy fiber (mf) terminals (A). In the LPS injected animal (B), BACE1 immunoreactivity is increased in the neuropil in the ipsilateral relative to contralateral cortex and hippocampal formation, especially apparent around the needle track (outline by arrows). At high magnification, swollen neurites are found in the cortex and hippocampal formation in the ipsilateral side, especially evident in the white matter, stratum radiatum (s.r.), stratum-lacunosum-moleculare (s.l.m.) and the striatum oriens (s.o.) that show little labeling in the normal conditions ((C), (D), (F)). Panels ((E), (F)) are taken from the section that is ~480 μm (12 × 30 μm) apart from (B), showing the distinct difference regarding the neuritic pathology between the two sides. The needle entry at the cortical surface is marked with an asterisk (*). Ctx: cortex (grey matter); DG: dentate gyrus; PC: parietal cortex; s.l.m.: stratum-lacunosum-moleculare; s.p.: stratum pyramidale. WM: white matter; Scale bar = 1 mm in (A) applying to (B); equivalent for 400 μm for ((C), (D)) and 200 μm for (E). sphericles and swollen processes were partially colocalized with the presynaptic marker synaptophysin (Figures 4(M)-(P)). In contrast, the BACE1 labeled swollen neurites did not colocalize with MAP2, which is clearly expressed in dendrites and somata of nearby pyramidal neurons (Figures 4(R)-(U)). 3.4. LPS Injection Induced Somotodendritic Changes in Cerebral Principal Neurons In our Golgi preparations, pyramidal neurons around layer III were consistently impregnated in all brain samples (Figure 5(A)). In the hippocampal proper, the curving area of CA3, whereby the s.p. continues dorsally from the X. H. Deng et al. 85 Figure 4. Confocal double immunofluorescent images showing LPS induced axonal pathology associated with increased labeling of amyloidogenic proteins. All images are taken from the CA3 area of the hippocampus ipsilateral to the intracerebral injection of PBS ((A)-(C)) or LPS ((E)-(U)). Antibody markers and color channels are as indicated. Panels ((A)-(C)) show double labeling of BACE1 and β-amyloid precursor protein (APP) in ipsilateral CA3 of the control animal, with the former expressed predominantly in the mossy fiber (mf) terminals, and the latter largely in somata of CA3 pyramidal neurons. Note that no abnormal neurites are present in the strum oriens (s.o.). Panels ((E)-(H)) illustrate the occurrence of BACE1/APP double labeled swollen neurites (examples are indicated by arrows) in the s.o. of the ipsilateral hippocampus of the LPS injected rat. Panels (I-L) show that these BACE1 positive neurites are locally associated with increased 4G8 labeling within (pointed by arrows) as well as outside (arrowheads) the swollen terminals in the cortex and CA1. Panels ((M)-(P)) show that a partial coexpression of synaptophysin (SYN) among the BACE1 labeled swollen neurites in the s.o. of the LPS injected ipsilateral hippocampus, which appear in yellow in the merged image (arrows, P). Panels (R-U) show that there is no colocalization of the microtubule associated protein-2 (MAP2) in the BACE1 labeled swollen neurites. MAP2 labeling is distinctly associated with the somata and dendrites of pyramidal neurons in the stratum pyramidale (s.p.). DAPI counterstain is included in panels (L) and (U), showing that the BACE1 labeled elements are not somatic. Scale bar = 200 μm in (A) applying to ((B), (C), (E)-(G) and (R)-(T)), equivalent to 100 μm for (I)-(K), (M)-(O)), 50 μm for ((D), (H), (U)) and 25 μm for ((L), (P)). CA1 direction and turns ventrally toward the dentate gyrus, could be defined systematically across brains. Therefore, we decided to use Golgi-stained pyramidal neurons in cortical layer III and around the dorsoventral turning area of CA3 for automated Neurolucida reconstruction and morphometry. Measurements from reconstructed layer III and CA1 pyramidal neurons in the ipsilateral hemisphere of the LPS and PBS treated brains X. H. Deng et al. 86 Figure 5. Quantitative Golgi study of somal and dendritic changes in cortical layer III pyramidal neurons in LPS-injected relative to PBS-injected rats. Panel (A) shows an example of Golgi stain in the parietal neocortex (ipsilateral to PBS injection), note that layer III pyramidal neurons are relatively well-labeled. Panels ((B), (C)) show representative Neurolucida reconstructions of impregnated layer III pyramidal neurons from a LPS (B) and a PBS (C) injected brains, with their apical and basal dendrites and dendritic branching points illustrated in color. Bar graphs ((D)-(G)) summarize the parameters obtained from 10 constructed neurons in each brain (n = 4/group). The means of somal perimeter and somal area show a trend of reduction in the LPS relative to PBS (control) groups, although the difference is not statistically significant (E). The means of total dendritic length tends to reduce in the LPS group relative to control, with significant difference for that of the basal dendritic tree (F). The branching nodes (points) are reduced in the LPS group on the basal dendritic tree (G). Overall spine density is significantly reduced on the dendritic tree of Golgi-impregnated cortical pyramidal neurons in the LPS group relative to control (H). Scale bar = 200 μm in (A). (10 cells per region per brain) were compared quantitatively (Figures 5(A)-(C) and Figures 6(A)-(D)). The somal perimeters of the layer III pyramidal neurons were comparable between the LPS (58.6 ± 7.4 µm, mean ± S.D., same format below) and PBS (61.7 ± 9.9 µm) groups (P > 0.05, two-tail paired t-test; same statistical test below) (Figure 5(D)). The mean somal area of layer III pyramidal neurons was reduced in the LPS (168.4 ± 21.6 µm2 ) relative to the PBS (216.5 ± 45.3 µm2 ) groups (P < 0.001). The total length of the dendritic processes of the layer III pyramidal neurons tended to be reduced in the LPS relative to PBS groups (Figure 5(E)). Thus, the total length of the apical dendrites was 647.4 ± 182.9 µm in the LPS group as compared to 834.9 ± 228.8 µm in the PBS group, with the means not statistically different (P > 0.05). The means of the total length of the basal dendrites were 711.4 ± 226.9 µm and 1122.0 ± 320.7 µm in the LPS and PBS groups, respectively, showing a significant difference (P < 0.001). The means of the branching points (nodes) on the apical dendrites were 7.0 ± 2.5 and 8.5 ± 2.4 in the LPS and the PBS groups, respectively, while they were 7.2 ± 1.3 X. H. Deng et al. 87 Figure 6. Quantitative Golgi study of somal and dendritic changes in in CA3 pyramidal neurons in LPS-injected relative to PBS-injected rat brains. Panel (A) shows an example of Golgi-stained CA3 pyramidal neurons located around dorsal to ventral turning area of the stratum pyramidale (s.r.). Basal dendrites in the stratum oriens (s.o.) and apical dendrites in the stratum radiatum (s.r.) are visible, with dendritic spines clearly labeled at high resolution (B). Panels ((C), (D)) show examples of Neurolucida reconstructions of CA3 pyramidal neurons from LPS (C) and a PBS (D) injected brains. The reduction in mean somal area (SA) in the LPS relative to PBS groups is statistically significant, whereas the reduction in the somal perimeter (SP) is not (E). The reduction in the total dendritic length in the LPS relative to PBS groups is significant for both the basal and apical trees (F). Scale bar = 50 μm in (A) and 5 μm for (B). and 10.6 ± 2.7 for the two groups on the basal dendrites. Statistical tests reported a significant difference between the two groups for the means of the basal dendritic but not the apical measurements (Figure 5(F)). The number of spines per unit length of dendrite was reduced significantly on the layer III pyramidal neurons in the LPS relative to PBS groups (Figure 5(G)). Data from the apical and basal dendrites combined, showed spine density to be 6.3 ± 0.9 vs. 8.0 ± 1.1 for the LPS relative to PBS groups (P < 0.0001). For the reconstructed CA3 pyramidal neurons (Figures 6(A)-(D)), both the somal perimeter and somal area of the pyramidal neurons tended to reduce in the LPS relative to PBS groups (Figure 6(E)). Thus, the mean somal perimeters were 47.8 ± 11.0 µm (mean ± S.D., same format below) and 67.8 ± 12.3 µm in the LPS and PBS treated animals, respectively (P > 0.05, two-tail paired t-test, same statistical test below). The reduction in the somal area of CA3 pyramidal neurons was significant (P < 0.001) in the LPS group (144.8 ± 40.4 µm2 ) compared to the PBS group (278.3 ± 60.2 µm2 ). There were significant decreases in the total lengths of the apical dendrites (320.0 ± 52.6 vs. 690.0 ± 63.6 µm; P < 0.005) and the basal dendrites (534.6 ± 70.6 vs. 967.0 ± 90.6 µm; P < 0.001) in the LPS compared to PBS groups (Figure 6(F)). The number of branching points on the apical dendrites (4.3 ± 2.1 vs. 8.3 ± 2.1; P < 0.001) and basal dendrites (3.9 ± 1.5 vs 7.2 ± 1.7; P < 0.001) of CA1 pyramidal neurons was significantly reduced in the LPS relative to PBS groups (Figure 6(G)). Finally, the density of spines on the apical and basal dendrites of the CA3 pyramidal neurons was significantly reduced (P < 0.001) in the LPS animals (10.5 ± 3.2) relative to PBS controls (18.0 ± 3.3) (Figure 6(H)). 4. Discussion 4.1. The LPS Model for Studying Chronic Neurodegeneration and Alzheimer’s Disease The LPS model is increasingly used in mechanistic and translational studies into chronic neurodegenerative dis X. H. Deng et al. 88 eases such as AD and PD [15] [16] [38] [44]-[50]. This relatively simple experimental approach offers yet a number of advantages in elucidating the neurobiology of aging and age-relative diseases. This model recapitulates many cellular/molecular events of neuroinflammation, which is considered an early, perhaps fundamental, causal factor in chronic neuronal and synaptic degeneration [8]-[14] [36]-[41]. The model also reproduces some defining clinical manifestations of human neurodegenerative diseases in rodents [44]-[46]. For instance, peripheral and central LPS administration induces learning/memory deficits in experimental animals analogous to AD-like cognitive decline [39]-[41]. Importantly, LPS treatment elicits neuropathological and behavioral changes in wild-type animals [47]-[50]. As neurodegenerative diseases occur mostly in a sporadic nature, the LPS model serves an ideal system to address pathogenic mechanisms underlying common human neurological disorders. Specifically, this study examined whether inflammation is a consequence or cause of dystrophic amyloidogenic neuritis. We have shown in a recent study that unilateral intracerebral injection of ~10 μg LPS is sufficient to induce spatial learning and memory impairments in adult rats. Upregulation of immunoinflammatory molecules including PirB and GFAP is evident in the LPS-treated brains, occurring largely in the ipsilateral cortex and hippocampal formation [41]. In the present study we observe that this same dose of LPS injection dramatically induces MHC II expression in the ipsilateral neocortex and hippocampal formation. This modulation appears to be largely associated with microglial activation, a key finding in the initial studies that establish this model of neurological diseases [36] [39]. Consistent with other reports [51], LPS-induced MHC II upregulation also appears to occur to a lesser extent in activated astrocytes that express high levels of GFAP. Thus, our data are consistent with the notion that intracranial LPS injection induces profound glial immunoinflammatory responses in the brain. 4.2. Axonal and Dendritic Pathology in LPS-Induced Neuroinflammation Besides glial responses, neuronal and synaptic alterations have been reported in rodent LPS models anatomically and electrophysiologically. Local LPS infusion causes increased cell/neuronal death in the entorhinal cortex [52] and substantia nigra [53] in rats as well as the hippocampus in ICR mice [54]. In BALB/c mice, the length and branching points of apical dendrites of CA1 pyramidal neurons are reduced in old but not young adults 72 hours following intraperitoneal LPS injection, while no change in spine density is found on these neurons [55]. In C57BL/6 mice, an increase in the density of thin spines on the dendrites of adult-born granule cells is found in the inner but not outer molecular layer 28 days following intrahippocampal LPS injection [56]. In adult rats, intrahippocampal LPS injection does not alter the intrinsic membrane properties, dendritic arborization or spine formation of the newly generated granule cells, but affect the overall network activity in the hippocampal neural circuitries [57]. LPS treatment can alter hippocampal synaptoplasticity in rats as measured by long-term potential and long-term depression [52] [58]-[60]. Here we demonstrate pre- and post-synaptic alterations among cortical and hippocampal neurons in LPS relative to PBS injected rats. Microscopically evident axonal pathology is featured by aberrant sprouting and swelling, and increased expression of APP, BACE1 and synaptophysin, but not MAP2. These abnormal axonal profiles seen following LPS treatment resemble the dystrophic neurites seen in models of injury that exhibit an increase in APP or A-beta [61] [62], although they do not arrange in typical rosette-like clusters as seen around established compact plaques [32]. Our Golgi data indicate that focal LPS injection causes a significant decrease in dendritic length and nodes on the basal tree as well as a reduction of spine density on the entire dendritic tree of layer III cortical principal neurons. The impact of focally injected LPS appears to be greater on CA3 pyramidale neurons, including significant reductions of somal size, lengths of basal and apical dendrites, branching points of the basal and apical dendrites, and overall spine density. The vulnerability of CA3 pyramidale neurons to LPS toxicity might relate to an intrinsic property of these cells in regard to synaptoplasticity. The dendrites and spines of CA3 pyramidal neurons belong to one of most plastic network systems in the brain—they receive major presynaptic inputs from the mossy fiber terminals that are constantly renewed in the process of adult neurogenesis of the dentate granule cells [63]. Alternatively, the observed variable somotodendritic changes between the cortical and hippocampal pyramidal neurons may reflect a potential spatial or dose-related effect by the injected toxin (i.e., the proximity to the injection site), which can be expected since the inflammation and BACE1 upregulation are not evident in the contralateral hemisphere. X. H. Deng et al. 89 4.3. Amyloid Pathogenesis in LPS-Induced Neuroinflammation LPS administration has been shown to influence amyloid pathogenesis via mixed cellular modulations in wild-type and transgenic rodents. Chronic intraventricular LPS infusion in rats induces intraneuronal Aβ immunoreactivity in the hippocampus [37]. Repeated intraperitoneal LPS injections in ICR mice result in increased intraneuronal and extracellular Aβ42 immunoreactivity in the cortex and hippocampus, with biochemical data indicating reduced α-secretase activity, increased γ-secretase activity, and elevated BACE1 protein and activity in tissue lysate [64]. In transgenic models of AD, LPS administration is shown to increase APP and Aβ immunoreactivity in neuronal somata [65], elevate γ-secretase activity [66] and accelerate plaque deposition [67]. However, other reports show reduced amyloid deposition via enhanced Aβ clearance by activated microglia after LPS administration [68] [69]. Being the obligatory enzyme for Aβ genesis, BACE1 is at the crossing point potentially linking cellular and signaling substrates to amyloid pathogenesis. BACE1 elevation is associated with axonal pathology during plaque development in the brains of transgenic AD models as well as aged and AD human subjects [32] [70]. As elaborated in the preceding section, we have identified axonal sprouting and swelling associated with enhanced APP, BACE1 and Aβ antibody reactivity in the ipsilateral cortex and hippocampal formation in LPS treated rat brains, especially evident around the needle track. This finding clearly indicates that chronic neuroinflammation can act as an inductive factor for amyloidogenic axonal pathology in wild-type adult mammalian brain. More basic questions can be asked as to why axonal sprouting/dystrophy, dendritic/spine degeneration and glial activation develop in partnership under neuroinflammatory conditions. One possibility is that the toxin interrupts axonal transport causing accumulation of the amyloidogenic proteins and other neuronal molecules [31]. This is consistent with the observation that the presynaptic marker synaptophysin is present only among a subset of the BACE1 immunoreactive swollen neurites. One may also speculate that this pathogenic correlation potentially involves the triadic organization of the synapse—presynaptic and postsynaptic terminals engulfed by glial processes. Neuroinflammation could initially induce some disengagement between the synaptic terminals and glial scaffold. For instance, inflammation-associated structural and functional changes in the glial cells may disrupt the fitness, integrity and plasticity of the pre- and post-synaptic terminals. This may eventually lead to major degenerative changes in the postsynaptic components, resulting in dendritic shrinkage and spine loss. The presynaptic components may also largely regress in the course of synaptic degeneration. However, some axon and presynaptic terminals might otherwise undergo an aggressive regenerative attempt because of the loss of appropriate postsynaptic targets. In this sense, the aberrant sprouting and dystrophy of the axonal terminals may be considered as a form of maladaptive neuronal regeneration, with the activated amyloidogenic machinery being a part of the its complex molecular dyshomeostasis. In summary, the present study demonstrates that chronic neuroinflammation induced by intracerebral LPS injection promotes the amyloidogenic cascade in dystrophic neurites by upregulation of BACE1 and APP in the brain. This modulation is evident in axon terminals that exhibit dystrophic-like morphology. LPS injection also induces degenerative changes in postsynaptic components manifested as dendritic shrinkage and spine loss among cortical and hippocampal pyramidal neurons. Together with the immunoinflammatory responses of the glial cells, the LPS model recapitulates multiple cellular and molecular deficits seen in aging and AD brains. The synaptic triad could be the initial site as well as center of neural degeneration and aberrant regeneration, with its progression ultimately leading to overt neuritic amyloid pathology and cognitive deficits. Acknowledgements This study was supported by the National Natural Science Foundation of China (#81171091, #81171160), Natural Science Foundation of Hunan Province (13JJ5016), Central South University (CSUZC2014035), Center for Alzheimer’s Disease and Related Disorders (P.R.P), and the Intramural research program of National Institute on Aging, NIH (Z01-IAAG000944-04 to H.C.). References [1] McGeer, E.G. and McGeer, P.L. (2010) Neuroinflammation in Alzheimer’s Disease and Mild Cognitive Impairment: A Field in Its Infancy. Journal of Alzheimer’s Disease, 19, 355-361. [2] Qian, L., Flood, P.M. and Hong, J.S. (2010) Neuroinflammation is a Key Player in Parkinson’s Disease and a Prime X. H. Deng et al. 90 Target for Therapy. Journal of Neural Transmission, 117, 971-979. http://dx.doi.org/10.1007/s00702-010-0428-1 [3] Broussard, G.J., Mytar, J., Li, R.C. and Klapstein, G.J. (2012) The Role of Inflammatory Processes in Alzheimer’s Disease. Inflammopharmacology, 20, 109-126. [4] Luessi, F., Siffrin, V. and Zipp, F. (2012) Neurodegeneration in Multiple Sclerosis: Novel Treatment Strategies. Expert Review of Neurotherapeutics, 12, 1061-1076. http://dx.doi.org/10.1586/ern.12.59 [5] Rao, J.S., Kellom, M., Kim, H.W., Rapoport, S.I. and Reese, E.A. (2012) Neuroinflammation and Synaptic Loss. Neurochemical Research, 37, 903-910. http://dx.doi.org/10.1007/s11064-012-0708-2 [6] Blandini, F. (2013) Neural and Immune Mechanisms in the Pathogenesis of Parkinson’s Disease. Journal of NeuroImmune Pharmacology, 8, 189-201. http://dx.doi.org/10.1007/s11481-013-9435-y [7] Breunig, J.J., Guillot-Sestier, M.V. and Town, T. (2013) Brain Injury, Neuroinflammation and Alzheimer’s Disease. Frontiers in Aging Neuroscience, 11, 26. [8] Enciu, A.M. and Popescu, B.O. (2013) Is There a Causal Link between Inflammation and Dementia? BioMed Research International, 2013, 316495. http://dx.doi.org/10.1155/2013/316495 [9] Ownby, R.L. (2010) Neuroinflammation and Cognitive Aging. Current Psychiatry Reports, 12, 39-45. http://dx.doi.org/10.1007/s11920-009-0082-1 [10] Pizza, V., Agresta, A., D’Acunto, C.W., Festa, M. and Capasso, A. (2011) Neuroinflammation and Ageing: Current Theories and an Overview of the Data. Reviews on Recent Clinical Trials, 6, 189-203. http://dx.doi.org/10.2174/157488711796575577 [11] Cagnin, A., Brooks, D.J., Kennedy, A.M., Gunn, R.N., Myers, R. and Turkheimer, F.E. (2001) In-Vivo Measurement of Activated Microglia in Dementia. Lancet, 358, 461-467. http://dx.doi.org/10.1016/S0140-6736(01)05625-2 [12] Butcher, S.K. and Lord, J.M. (2004) Stress Responses and Innate Immunity: Aging as a Contributory Factor. Aging Cell, 3, 151-160. http://dx.doi.org/10.1111/j.1474-9728.2004.00103.x [13] Eikelenboom, P., van Exel, E., Hoozemans, J.J., Veerhuis, R., Rozemuller, A.J. and van Gool, W.A. (2010) Neuroinflammation—An Early Event in Both the History and Pathogenesis of Alzheimer’s Disease. Neurodegenerative Diseases, 7, 38-41. http://dx.doi.org/10.1159/000283480 [14] Wang, X., Wang, W., Li, L., Perry, G., Lee, H.G. and Zhu, X. (2013) Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochimica et Biophysica Acta, S0925-4439(13)00323-2. [15] Katafuchi, T., Ifuku, M., Mawatari, S., Noda, M., Miake, K., Sugiyama, M. and Fujino, T. (2012) Effects of Plasmalogens on Systemic Lipopolysaccharide-Induced Glial Activation and β-Amyloid Accumulation in Adult Mice. Annals of the New York Academy of Sciences, 1262, 85-92. http://dx.doi.org/10.1111/j.1749-6632.2012.06641.x [16] Krstic, D., Madhusudan, A., Doehner, J., Vogel, P., Notter, T., Imhof, C., Manalastas, A., Hilfiker, M., Pfister, S., Schwerdel, C., Riether, C., Meyer, U. and Knuesel, I. (2012) Systemic Immune Challenges Trigger and Drive AlzheimerLike Neuropathology in Mice. Journal of Neuroinflammation, 9, 151. http://dx.doi.org/10.1186/1742-2094-9-151 [17] Kraft, A.W., Hu, X., Yoon, H., Yan, P., Xiao, Q., Wan, Y., Gil, S.C., Brown, J., Wilhelmsson, U., Restivo, J.L., Cirrito, J.R., Holtzman, D.M., Kim, J., Pekny, M. and Lee, J.M. (2013) Attenuating Astrocyte Activation Accelerates Plaque Pathogenesis in APP/PS1 Mice. FASEB Journal, 27, 187-198. http://dx.doi.org/10.1096/fj.12-208660 [18] Medeiros, R. and LaFerla, F.M. (2013) Astrocytes: Conductors of the Alzheimer Disease Neuroinflammatory Symphony. Experimental Neurology, 239, 133-138. http://dx.doi.org/10.1016/j.expneurol.2012.10.007 [19] Zhao, J., O’Connor, T. and Vassar, R. (2011) The Contribution of Activated Astrocytes to Aβ Production: Implications for Alzheimer’s Disease Pathogenesis. Journal of Neuroinflammation, 8, 150. http://dx.doi.org/10.1186/1742-2094-8-150 [20] Sondag, C.M., Dhawan, G. and Combs, C.K. (2009) Beta Amyloid Oligomers and Fibrils Stimulate Differential Activation of Primary Microglia. Journal of Neuroinflammation, 6, 1. http://dx.doi.org/10.1186/1742-2094-6-1 [21] Maezawa, I., Zimin, P.I., Wulff, H. and Jin, L.W. (2011) Amyloid-Beta Protein Oligomer at Low Nanomolar Concentrations Activates Microglia and Induces Microglial Neurotoxicity. Journal of Biological Chemistry, 286, 3693-3706. http://dx.doi.org/10.1074/jbc.M110.135244 [22] DeKosky, S.T. and Scheff, S.W. (1990) Synapse Loss in Frontal Cortex Biopsies in Alzheimer’s Disease: Correlation with Cognitive Severity. Annals of Neurology, 27, 457-464. http://dx.doi.org/10.1002/ana.410270502 [23] Terry, R.D., Masliah, E., Salmon, D.P., Butters, N., Deteresa, R., Hill, R., Hansen, L.A. and Katzman, R. (1991) Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Annals of Neurology, 30, 572-580. http://dx.doi.org/10.1002/ana.410300410 [24] Masliah, E., Mallory, M., Alford, M., DeTeresa, R., Hansen, L.A., McKeel Jr., D.W. and Morris, J.C. (2001) Altered Expression of Synaptic Proteins Occurs Early during Progression of Alzheimer’s Disease. Neurology, 56, 127-129. http://dx.doi.org/10.1212/WNL.56.1.127 X. H. Deng et al. 91 [25] Heinonen, O., Soininen, H., Sorvari, H., Kosunen, O., Paljarvi, L., Koivisto, E. and Riekkinen, P.J. (1995) Loss of Synaptophysin-Like Immunoreactivity in the Hippocampal Formation Is an Early Phenomenon in Alzheimer’s Disease. Neuroscience, 64, 375-384. http://dx.doi.org/10.1016/0306-4522(94)00422-2 [26] Reddy, P.H., Mani, G., Park, B.S., Jacques, J., Murdoch, G., Whetsell Jr., W., Kaye, J. and Manczak, M. (2005) Differential Loss of Synaptic Proteins in Alzheimer’s Disease: Implications for Synaptic Dysfunction. Journal of Alzheimer’s Disease, 7, 103-117. [27] Scheff, S.W., Price, D.A., Schmitt, F.A., Scheff, M.A. and Mufson, E.J. (2011) Synaptic Loss in the Inferior Temporal Gyrus in Mild Cognitive Impairment and Alzheimer’s Disease. Journal of Alzheimer’s Disease, 24, 547-557. [28] D’Amelio, M. and Rossini, P.M. (2012) Brain Excitability and Connectivity of Neuronal Assemblies in Alzheimer’s Disease: From Animal Models to Human Findings. Progress in Neurobiology, 99, 42-60. http://dx.doi.org/10.1016/j.pneurobio.2012.07.001 [29] Berchtold, N.C., Coleman, P.D., Cribbs, D.H., Rogers, J., Gillen, D.L. and Cotman, C.W. (2013) Synaptic Genes Are Extensively Downregulated across Multiple Brain Regions in Normal Human Aging and Alzheimer’s Disease. Neurobiology of Aging, 34, 1653-1661. http://dx.doi.org/10.1016/j.neurobiolaging.2012.11.024 [30] Knobloch, M. and Mansuy, I.M. (2008) Dendritic Spine Loss and Synaptic Alterations in Alzheimer’s Disease. Molecular Neurobiology, 37, 73-82. http://dx.doi.org/10.1007/s12035-008-8018-z [31] Kanaan, N.M., Pigino, G.F., Brady, S.T., Lazarov, O., Binder, L.I. and Morfini, G.A. (2013) Axonal Degeneration in Alzheimer’s Disease: When Signaling Abnormalities Meet the Axonal Transport System. Experimental Neurology, 246, 44-53. http://dx.doi.org/10.1016/j.expneurol.2012.06.003 [32] Yan, X.X., Ma, C., Gai, W.P., Cai, H. and Luo, X.G. (2014) Can BACE1 Inhibition Mitigate Early Axonal Pathology in Neurological Diseases? Journal of Alzheimer’s Disease, 38, 705-718. [33] Zhang, X.M., Cai, Y., Xiong, K., Cai, H., Luo, X.G., Feng, J.C., Clough, R.W., Struble, R.G., Patrylo, P.R. and Yan, X.X. (2009) β-Secretase-1 Elevation in Transgenic Mouse Models of Alzheimer’s Disease Is Associated with Synaptic/ Axonal Pathology and Amyloidogenesis: Implications for Neuritic Plaque Development. European Journal of Neuroscience, 30, 2271-2283. http://dx.doi.org/10.1111/j.1460-9568.2009.07017.x [34] Cai, Y., Zhang, X.M., Macklin, L.N., Cai, H., Luo, X.G., Oddo, S., Laferla, F.M., Struble, R.G., Rose, G.M., Patrylo, P.R. and Yan, X.X. (2012) BACE1 Elevation Is Involved in Amyloid Plaque Development in the Triple Transgenic Model of Alzheimer’s Disease: Differential Aβ Antibody Labeling of Early-Onset Axon Terminal Pathology. Neurotoxicity Research, 21, 160-174. http://dx.doi.org/10.1007/s12640-011-9256-9 [35] Li, J.M., Xue, Z.Q., Deng, S.H., Luo, X.G., Patrylo, P.R., Rose, G.W., Cai, H., Cai, Y. and Yan, X.X. (2013) Amyloid Plaque Pathogenesis in 5XFAD Mouse Spinal Cord: Retrograde Transneuronal Modulation after Peripheral Nerve Injury. Neurotoxicity Research, 24, 1-14. http://dx.doi.org/10.1007/s12640-012-9355-2 [36] Xu, J. and Ling, E.A. (1994) Upregulation and Induction of Surface Antigens with Special Reference to MHC Class II Expression in Microglia in Postnatal Rat Brain Following Intravenous or Intraperitoneal Injections of Lipopolysaccharide. Journal of Anatomy, 184, 285-296. [37] Hauss-Wegrzyniak, B., Dobrzanski, P., Stoerh, J.D. and Wenk, G.L. (1998) Chronic Neuroinflammation in Rats Reproduces Components of the Neurobiology of Alzheimer’s Disease. Brain Research, 780, 294-303. http://dx.doi.org/10.1016/S0006-8993(97)01215-8 [38] Sugaya, K., Chou, S., Xu, S.J. and McKinney, M. (1998) Indicators of Glial Activation and Brain Oxidative Stress after Intraventricular Infusion of Endotoxin. Molecular Brain Research, 58, 1-9. http://dx.doi.org/10.1016/S0169-328X(97)00365-3 [39] Tanaka, S., Ide, M., Shibutani, T., Ohtaki, H., Numazawa, S., Shioda, S. and Yoshida, T. (2006) LipopolysaccharideInduced Microglial Activation Induces Learning and Memory Deficits without Neuronal Cell Death in Rats. Journal of Neuroscience Research, 83, 557-566. http://dx.doi.org/10.1002/jnr.20752 [40] Lee, J.W., Lee, Y.K., Yuk, D.Y., Choi, D.Y., Ban, S.B., Oh, K.W. and Hong, J.T. (2008) Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment through Enhancement of Beta-Amyloid Generation. Journal of Neuroinflammation, 5, 37. http://dx.doi.org/10.1186/1742-2094-5-37 [41] Deng, X.H., Ai, W.M., Lei, D.L., Luo, X.G., Yan, X.X. and Li, Z. (2012) Lipopolysaccharide Induces Paired Immunoglobulin-Like Receptor B (PirB) Expression, Synaptic Alteration, and Learning-Memory Deficit in Rats. Neuroscience, 209, 161-170. http://dx.doi.org/10.1016/j.neuroscience.2012.02.022 [42] Laird, F.M., Cai, H., Savonenko, A.V., Farah, M.H., He, K., Melnikova, T., Wen, H., Chiang, H.C., Xu, G., Koliatsos, V.E., Borchelt, D.R., Price, D.L., Lee, H.K. and Wong, P.C. (2005) BACE1, a Major Determinant of Selective Vulnerability of the Brain to Amyloid-β Amyloidogenesis, Is Essential for Cognitive, Emotional, and Synaptic Functions. Journal of Neuroscience, 25, 11693-11709. http://dx.doi.org/10.1523/JNEUROSCI.2766-05.2005 X. H. Deng et al. 92 [43] Xiong, K., Cai, H., Luo, X.G., Struble, R.G., Clough, R.W. and Yan, X.X. (2007) Mitochondrial Respiratory Inhibition and Oxidative Stress Elevate β-Secretase (BACE1) Proteins and Activity in Vivo in the Rat Retina. Experimental Brain Research, 181, 435-446. http://dx.doi.org/10.1007/s00221-007-0943-y [44] Dutta, G., Zhang, P. and Liu, B. (2008) The Lipopolysaccharide Parkinson’s Disease Animal Model: Mechanistic Studies and Drug Discovery. Fundamental & Clinical Pharmacology, 22, 453-464. http://dx.doi.org/10.1111/j.1472-8206.2008.00616.x [45] Lee, D.C., Rizer, J., Selenica, M.L., Reid, P., Kraft, C., Johnson, A., Blair, L., Gordon, M.N., Dickey, C.A. and Morgan, D. (2010) LPS-Induced Inflammation Exacerbates Phospho-Tau Pathology in rTg4510 Mice. Journal of Neuroinflammation, 7, 56. http://dx.doi.org/10.1186/1742-2094-7-56 [46] Hoban, D.B., Connaughton, E., Connaughton, C., Hogan, G., Thornton, C., Mulcahy, P., Moloney, T.C. and Dowd, E. (2013) Further Characterisation of the LPS Model of Parkinson’s Disease: A Comparison of Intra-Nigral and IntraStriatal Lipopolysaccharide Administration on Motor Function, Microgliosis and Nigrostriatal Neurodegeneration in the Rat. Brain, Behavior, and Immunity, 27, 91-100. http://dx.doi.org/10.1016/j.bbi.2012.10.001 [47] Ifuku, M., Katafuchi, T., Mawatari, S., Noda, M., Miake, K., Sugiyama, M. and Fujino, T. (2012) Anti-Inflammatory/ Anti-Amyloidogenic Effects of Plasmalogens in Lipopolysaccharide-Induced Neuroinflammation in Adult Mice. Journal of Neuroinflammation, 9, 197. http://dx.doi.org/10.1186/1742-2094-9-197 [48] Martin, S.A., Pence, B.D., Greene, R.M., Johnson, S.J., Dantzer, R., Kelley, K.W. and Woods, J.A. (2013) Effects of Voluntary Wheel Running on LPS-Induced Sickness Behavior in Aged Mice. Brain, Behavior, and Immunity, 29, 113- 123. http://dx.doi.org/10.1016/j.bbi.2012.12.014 [49] Samanani, S., Mishra, M., Silva, C., Verhaeghe, B., Wang, J., Tong, J. and Yong, V.W. (2013) Screening for Inhibitors of Microglia to Reduce Neuroinflammation. CNS & Neurological Disorders-Drug Targets, 12, 741-749. http://dx.doi.org/10.2174/18715273113126660177 [50] Asti, A. and Gioglio, L. (2014) Can a Bacterial Endotoxin Be a Key Factor in the Kinetics of Amyloid Fibril Formation? Journal of Alzheimer’s Disease, 39, 169-179. [51] Morga, E., Faber, C. and Heuschling, P. (1999) Regional Heterogeneity of the Astroglial Immunoreactive Phenotype: Effect of Lipopolysaccharide. Journal of Neuroscience Research, 57, 941-952. http://dx.doi.org/10.1002/(SICI)1097-4547(19990915)57:6<941::AID-JNR20>3.0.CO;2-Z [52] Hauss-Wegrzyniak, B., Lynch, M.A., Vraniak, P.D. and Wenk, G.L. (2002) Chronic Brain Inflammation Results in Cell Loss in the Entorhinal Cortex and Impaired LTP in Perforant Path-Granule Cell Synapses. Experimental Neurology, 176, 336-341. http://dx.doi.org/10.1006/exnr.2002.7966 [53] Burguillos, M.A., Hajji, N., Englund, E., Persson, A., Cenci, A.M., Machado, A., Cano, J., Joseph, B. and Venero, J.L. (2011) Apoptosis-Inducing Factor Mediates Dopaminergic Cell Death in Response to LPS-Induced Inflammatory Stimulus: Evidence in Parkinson’s Disease Patients. Neurobiology of Disease, 41, 177-188. http://dx.doi.org/10.1016/j.nbd.2010.09.005 [54] Lee, J.W., Lee, Y.K., Yuk, D.Y., Choi, D.Y., Ban, S.B., Oh, K.W. and Hong, J.T. (2008) Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment through Enhancement of Beta-Amyloid Generation. Journal of Neuroinflammation, 5, 37. http://dx.doi.org/10.1186/1742-2094-5-37 [55] Richwine, A.F., Parkin, A.O., Buchanan, J.B., Chen, J., Markham, J.A., Juraska, J.M. and Johnson, R.W. (2008) Architectural Changes to CA1 Pyramidal Neurons in Adult and Aged Mice after Peripheral Immune Stimulation. Psychoneuroendocrinology, 33, 1369-1377. http://dx.doi.org/10.1016/j.psyneuen.2008.08.003 [56] Chugh, D., Nilsson, P., Afjei, S.A., Bakochi, A. and Ekdahl, C.T. (2013) Brain Inflammation Induces Post-Synaptic Changes during Early Synapse Formation in Adult-Born Hippocampal Neurons. Experimental Neurology, 250, 176- 188. http://dx.doi.org/10.1016/j.expneurol.2013.09.005 [57] Jakubs, K., Bonde, S., Iosif, R.E., Ekdahl, C.T., Kokaia, Z., Kokaia, M. and Lindvall, O. (2008) Inflammation Regulates Functional Integration of Neurons Born in Adult Brain. Journal of Neuroscience, 28, 12477-12488. http://dx.doi.org/10.1523/JNEUROSCI.3240-08.2008 [58] Commins, S., O’Neill, L.A. and O’Mara, S.M. (2001) The Effects of the Bacterial Endotoxin Lipopolysaccharide on Synaptic Transmission and Plasticity in the CA1-Subiculum Pathway in Vivo. Neuroscience, 102, 273-280. http://dx.doi.org/10.1016/S0306-4522(00)00498-X [59] Jo, J.H., Park, E.J., Lee, J.K., Jung, M.W. and Lee, C.J. (2001) Lipopolysaccharide Inhibits Induction of Long-Term Potentiation and Depression in the Rat Hippocampal CA1 Area. European Journal of Pharmacology, 422, 69-76. http://dx.doi.org/10.1016/S0014-2999(01)01075-5 [60] Min, S.S., Quan, H.Y., Ma, J., Lee, K.H., Back, S.K., Na, H.S., Han, S.H., Yee, J.Y., Kim, C., Han, J.S. and Seol, G.H. (2009) Impairment of Long-Term Depression Induced by Chronic Brain Inflammation in Rats. Biochemical and Biophysical Research Communications, 383, 93-97. http://dx.doi.org/10.1016/j.bbrc.2009.03.133 X. H. Deng et al. 93 [61] Gentleman, S.M., Nash, M.J., Sweeting, C.J., Graham, D.I. and Roberts, G.W. (1993) β-Amyloid Precursor Protein (βAPP) as a Marker for Axonal Injury after Head Injury. Neuroscience Letters, 160, 139-144. http://dx.doi.org/10.1016/0304-3940(93)90398-5 [62] Chen, X.H., Johnson, V.E., Uryu, K., Trojanowski, J.Q. and Smith, D.H. (2009) A Lack of Amyloid Beta Plaques Despite Persistent Accumulation of Amyloid β in Axons of Long-Term Survivors of Traumatic Brain Injury. Brain Pathology, 19, 214-223. http://dx.doi.org/10.1111/j.1750-3639.2008.00176.x [63] Mongiat, L.A. and Schinder, A.F. (2011) Adult Neurogenesis and the Plasticity of the Dentate Gyrus Network. European Journal of Neuroscience, 33, 1055-1061. http://dx.doi.org/10.1111/j.1460-9568.2011.07603.x [64] Lee, J.W., Lee, Y.K., Yuk, D.Y., Choi, D.Y., Ban, S.B., Oh, K.W. and Hong, J.T. (2008) Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment through Enhancement of Beta-Amyloid Generation. Journal of Neuroinflammation, 5, 37. http://dx.doi.org/10.1186/1742-2094-5-37 [65] Sheng, J.G., Bora, S.H., Xu, G., Borchelt, D.R., Price, D.L. and Koliatsos, V.E. (2003) Lipopolysaccharide-InducedNeuroinflammation Increases Intracellular Accumulation of Amyloid Precursor Protein and Amyloid β Peptide in APPswe Transgenic Mice. Neurobiology of Disease, 14, 133-145. http://dx.doi.org/10.1016/S0969-9961(03)00069-X [66] Joshi, Y.B., Giannopoulos, P.F., Chu, J. and Praticò, D. (2014) Modulation of Lipopolysaccharide-Induced Memory Insult, γ-Secretase, and Neuroinflammation in Triple Transgenic Mice by 5-Lipoxygenase. Neurobiology of Aging, 35, 1024-1031. http://dx.doi.org/10.1016/j.neurobiolaging.2013.11.016 [67] Qiao, X., Cummins, D.J. and Paul, S.M. (2001) Neuroinflammation-Induced Acceleration of Amyloid Deposition in the APPV717F Transgenic Mouse. European Journal of Neuroscience, 14, 474-482. http://dx.doi.org/10.1046/j.0953-816x.2001.01666.x [68] DiCarlo, G., Wilcock, D., Henderson, D., Gordon, M. and Morgan, D. (2001) Intrahippocampal LPS Injections Reduce Aβ Load in APP+PS1 Transgenic Mice. Neurobiology of Aging, 22, 1007-1012. http://dx.doi.org/10.1016/S0197-4580(01)00292-5 [69] Herber, D.L., Mercer, M., Roth, L.M., Symmonds, K., Maloney, J., Wilson, N., Freeman, M.J., Morgan, D. and Gordon, M.N. (2007) Microglial Activation Is Required for Aβ Clearance after Intracranial Injection of Lipopolysaccharide in APP Transgenic Mice. Journal of Neuroimmune Pharmacology, 2, 222-231. http://dx.doi.org/10.1007/s11481-007-9069-z [70] Kandalepas, P.C., Sadleir, K.R., Eimer, W.A., Zhao, J., Nicholson, D.A. and Vassar, R. (2013) The Alzheimer’s β-Secretase BACE1 Localizes to Normal Presynaptic Terminals and to Dystrophic Presynaptic Terminals Surrounding Amyloid Plaques. Acta Neuropathologica, 126, 329-352. http://dx.doi.org/10.1007/s00401-013-1152-3 Scientific Research Publishing (SCIRP) is one of the largest Open Access journal publishers. It is currently publishing more than 200 open access, online, peer-reviewed journals cov
Lipolysaccharide-Induced Neuroinflammation Is Associated with Alzheimer-Like Amyloidogenic Axonal Pathology and Dendritic Degeneration in Rats Xiaohua Deng1*, Meili Li1*, Weiming Ai1,2, Lixin He1,3, Dahua Lu1, Peter R. Patrylo4, Huaibin Cai5, Xuegang Luo1, Zhiyuan Li1, Xiao-Xin Yan1# 1 Department of Anatomy and Neurobiology, Central South University School of Basic Medical Science, Changsha, China 2 Department of Nursing in Internal Medicine, School of Nursing, Xiangtan Vocational and Technical College, Xiangtan, China 3 Department of Anatomy and Physiology, School of Nursing, Xiangtan Vocational and Technical College, Xiangtan, China 4 Departments of Physiology, Anatomy and the Center for Integrated Research in Cognitive and Neural Sciences, Southern Illinois University Carbondale, Carbondale, USA 5 Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, USA Email: # yanxiaoxin@csu.edu.cn Received 9 May 2014; revised 31 May 2014; accepted 7 June 2014 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Chronic neuroinflammation is thought to play an etiological role in Alzheimer’s disease (AD) which is characterized pathologically by amyloid and tau formation, as well as neuritic dystrophy and synaptic degeneration. The causal relationship between these pathological events is a topic of ongoing research and discussion. Recent data from transgenic AD models point to a tight spatiotemporal link between neuritic and amyloid pathology, with the obligatory enzyme for β-amyloid (Aβ) production, namely β-secretase-1 (BACE1), being overexpressed in axon terminals undergoing dystrophic change. However, the axonal pathology inherent with BACE1 elevation seen in transgenic AD mice may be secondary to increased soluble Aβ in these genetically modified animals. Further, it is unclear whether the inflammation seen in AD is the result of , or the cause of * Authors contributed to this work equally. # Corresponding author. X. H. Deng et al. 79 neuritic dystrophy. Here we explored the occurrence of AD-like axonal and dendritic pathology in adult rat brains affected by LPS-induced chronic neuroinflammation. Unilateral intracerebral LPS injection induced prominent inflammatory response in glial cells in the ipsilateral cortex and hippocampal formation. BACE1 protein levels were elevated in the ipsilateral hippocampal lysates in the LPS-treated animals relative to controls. BACE1 immunoreactive dystrophic axons appeared in the LPS-treated ipsilateral cortex and hippocampal formation, colocalizing with increased β-amyloid precursor protein and Aβ antibody (4G8) immunolabeling. Quantitative Golgi studies revealed reduction of dendritic branching points and spine density on cortical layer III and hippocampal CA3 pyramidal neurons in the LPS-treated ipsilateral cerebrum. These findings suggest that Alzheimer-like amyloidogenic axonal pathology and dendritic degeneration occur in wildtype mammalian brain in partnership with neuroinflammation following LPS injection. Keywords Amyloid Pathogenesis, Neuritic Dystrophy, Neurodegeneration, Neuroplasticity, Synaptic Pathology 1. Introduction Neuroinflammation has been linked to many neuropsychiatric disorders, including neurodegenerative diseases such as Alzheimer’s diseases (AD), Parkinson’s disease (PD), multiple sclerosis and traumatic brain injury [1]- [8]. Aging is a major risk factor for many age-related diseases, and is associated with a certain degree of chronic inflammation [9] [10]. In general, chronic inflammation is considered to mount lasting stress on neurons and synapses, and may lead to brain dysfunction, including cognitive deficits [5] [11]-[14]. The causal relationship between chronic inflammation and some of the hallmark pathological lesions in neurological diseases is under intensive investigation. For example, oxidative or inflammatory stress is suggested to promote cerebral amyloid pathology via increased production and/or impaired clearance of Aβ, involving both neurons and glial cells [15]-[19]. Aβ products including soluble and aggregated variants may also act as proinflammatory factors [20] [21]. Loss of synapses and their connectivity best correlate with cognitive deficits in AD [22]-[29]. The density of dendritic spines appears to be reduced readily at prodromal stages of the disease [30]. Besides synaptic degeneration, axonal elements including presynaptic terminals undergo aberrant sprouting and dystrophic expansion [30]-[32]. Recent data from transgenic AD models, nonhuman primates and human subjects show that upregulation of the amyloidogenic proteins, especially the rate-limiting enzyme β-secretase-1 (BACE1), appears to be an molecular cascade tightly associated with axonal sprouting and dystrophy, suggestive of a driving role for amyloidogenic axonal pathology in plaque formation [33]-[35]. The bacterial endotoxin lipopolysaccharide (LPS) can induce chronic neuroinflammation in rodents [36]-[38]. LPS administration also causes learning and memory deficits in the animals, providing an excellent model system for studying cognitive dysfunction associated with chronic neuroinflammation [39]-[41]. Neuroinflammation is considered to play an early or inductive role in the development of AD pathologies, although the anatomic evidence remains to be better formulated. Therefore, the present study was set to address whether intracerebral LPS injection in adult rats may induce axonal and dendritic pathologies similar to that seen in AD [32]. Specifically, we aimed to identify whether dystrophic axonal pathology inherent with amyloidogenic modulation and degenerative dendritic/spine changes occur on cerebral principal neurons in LPS-treated adult rats. 2. Materials and Methods 2.1. Animals and Intracerebral Injection In-house bred male adult Sprague-Dawley rats (n = 24) weighing ~200 g (Animal Center of Central South University) were used. Rats were maintained in temperature (20˚C - 26˚C), humidity (30% - 60%) and lighting (12:12 hours light/dark cycle) controlled rooms, with food and water freely available. For intracerebral injections, rats were placed in a stereotaxic apparatus under sodium pentobarbital anesthesia (50 mg/kg, i.p.). LPS X. H. Deng et al. 80 from Escherichia coli serotype 055:B5 (L2637, Sigma-Aldrich, St. Louis, MO, USA) was dissolved (2.5 µg/µl) in sterile phosphate-buffered saline (PBS, 0.01 M, pH 7.2) (vehicle) [41]. LPS (10 µg in 4 µl) was injected through the neocortex into the right hippocampus with a microsyringe in each animal (n = 12), using the following coordinates: 3.5 mm lateral to the sagittal cranial suture; 5 mm caudal to the bregma and 3 mm below the dura mater. Controls (n = 12) were injected with the same amount of PBS. Brain examination was carried out 30 days post intracortical injection. The experimental protocol was approved by the Animal Care and Use Committee of the Central South University and were in compliance with NIH guidelines. 2.2. Immunoblot Hippocampi ipsilateral to the injection side were dissected out following decapitation (n = 4/group). Tissue was weighed and homogenized in a commercial protein extraction buffer at 1 to 10 w/v ratio (CW0883, Kangwei Century Company, Beijing), followed by centrifuge at 15,000 g. Protein concentrations in the supernatants were determined by DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA). A total of 25 µg protein from each sample was separated electrophoretically in 10% SDS-polyacrylamide gel and transferred to polyvinylidene fluoride membrane. Membranes were blocked with 1% non-fat milk and 5% bovine serum albumin (BSA) in 0.1 M Tris-buffered saline containing 0.1% Tween-20 (TBS-T) for 2 hours at room temperature. Membranes were then incubated overnight at 4˚C in the same buffer containing rabbit anti-BACE1 (1:2000) [33] [34] [42] [43], rabbit anti-glial fibrillary acidic protein (GFAP) (G9269, Sigma-Aldrich, St Louis, MO, USA, 1:4000) or rabbit anti-β-tubulin (Sigma-Aldrich, T2200, 1:10000). Membranes were washed thoroughly with TBS-T, then were incubated for 2 hours at room temperature with horseradish peroxidase-conjugated goat anti-rabbit IgGs (1:20,000; Bio-Rad Laboratories). Bound antibodies were detected by enhanced chemiluminescence (ECL kit, GE Healthcare Life Sciences, Piscataway, NJ, USA). The membranes were exposed to X-ray films developed subsequently in a darkroom. The films were scanned, with optical density (OD) of immunoblot bands measured using Image-J, followed by standardization to the internal references. 2.3. Immunohistochemistry Rats were perfused transcardially with PBS followed by 4% phosphate-buffered (0.1M) paraformaldehyde (pH 7.4). Brains were removed and postfixed for 12 hours and transferred into 30% sucrose for cryoprotection. Thirty micrometer-thick coronal sections were cut in a cryostat, with 12 sets of sections collected for cresyl violet stain and for immunohistochemical studies. For immunohistochemistry with the avidin-biotin complex (ABC) method, free-floating sections were soaked in 1% H2O2 in PBS for 30 minutes to diminish endogenous peroxidase activity, and preincubated in PBS buffer containing 0.2% Triton X-100 and normal horse serum for 1 hour. The sections were then reacted with one of the following primary antibodies: 1) Mouse monoclonal antibody against major histocompatibility complex class II molecules (MHC-II) at 1:1000 (ab55152, Abcam, Cambridge, MA, USA); 2) Rabbit anti-human BACE1 at 1:2000; 3) mouse anti-β-amyloid precursor protein (APP) monoclonal antibody 22C11 (MAB348, EMD Millipore, Billerica, MA, USA, 1:4000); 4) mouse anti-Aβ17-24 monoclonal antibody 4G8 (#39240, Signet, Dedham, USA, 1:4000); 5) rabbit anti-GFAP (G9269, Sigma-Aldrich, 1:2000). The sections were further reacted with a biotinylated pan-specific secondary antibody (BA-1300, Vector Laboratories, Burlingame, CA, USA, 1:400) for 2 hours, and subsequently with the ABC reagents (PK-6100, Vector Laboratories, 1:400) for one hour. Immunoreactivity was visualized in 0.05% 3,3’-diaminobenzidine (DAB) and 0.003% H2O2. Selected sections were processed for double immunofluorescence beginning with blocking nonspecific reactivity by incubation in PBS buffer containing 5% normal donkey serum. Sections were further reacted overnight at 4˚C with a pair of primary antibodies raised in different species in PBS containing 0.2% Triton X-100 and the blocking serum. The antibody pairs included: 1) mouse anti-MCH-II (1:1000) and rabbit anti-CD11b (MABT149, EMD Millipore, 1:1000); 2) mouse anti-MCH-II and rabbit anti-GFAP (G9269, 1:2000); 3) rabbit anti-BACE1 and mouse anti-APP 22C11; 4) rabbit anti-BACE1 and mouse anti-Aβ 4G8; 5) rabbit anti-BACE1 and mouse anti-synaptophysin (MAB329, EMD Millipore, 1:4000); 6) rabbit anti-BACE1 and mouse anti-microtubule associated protein-2 (MAP2) (M9942, Sigma-Aldrich, 1:2000). On the second day, the sections were rinsed with PBS and incubated at room temperature for 2 hours with Alexa Fluor® 488 and Alexa Fluor® 594 conjugated donkey anti-mouse and anti-rabbit IgGs (1:200, Invitrogen, Carlsbad, CA, USA). Sections were then counter-stained with bisbenzimide (Hoechst 33342, 1:50000, Catalog #B2261, Sigma-Aldrich), washed thoroughly, and mounted with anti-fading medium before microscopic examination. X. H. Deng et al. 81 2.4. Rapid Golgi-Cox Stain Brains were removed following a vascular rinse with PBS. Blocks containing the middle 1/3 cerebrum of both hemispheres were rinsed briefly in double distilled water, and processed with the FD Rapid Golgi stain TM Kit (FD Neuro Technologies, Ellicott City, MD) following the manufacturer’s instruction. The brain blocks were immersed in freshly made mixture of Solutions A and B (1:1) in the dark at room temperature for 2 weeks, and then in Solution C at 4˚C in the dark for 3 days. After silver impregnation, the blocks were cut slowly into frontal sections at 100 μm thickness in a vibratome. Sections were collected alternatively in 10 sets in Solution C, mounted on gelatin-coated microslides, dehydrated through ascending concentrations of ethanol, cleared in xylene, and sealed with PermountTM mounting medium. 2.5. Imaging and Data Analysis An Olympus (BX53) microscope equipped with imaging system (CellSens Standard, Olympus) was used for examining sections stained with the ABC and fluorescent methods. Double immunofluorescence was also imaged on a confocal microscope (Nikon, DIGITAL ECLIPSE C1 plus, 5 μm thickness scan). Immunolabeling in sections around the level of injection in the rostrocaudal dimension was comparatively examined between the ipsilateral and contralateral cerebral hemispheres, using the needle track as a reference. Golgi-impregnated sections were examined on a Zeiss Axioplan microscope equipped with the Neurolucida and a high-resolution motorized stage for 3D neuronal reconstruction (MicroBrightField China). Two sections nearest to the injection coordinates (in the rostrocaudal dimension) were selected from each brain for neuronal morphometric analysis. Golgi-stained pyramidal neurons that met the following criteria were selected for reconstruction: 1) they were located in layer III of the parietal cortex overlying the mid-hippocampus and in the middle portion of CA3 (i.e., around the dorsal to ventral turning area); 2) they were among the labeled cells with the widest dendritic field by overall visual judgment; 3) they were well separated from other impregnated cells such that their dendritic tree was not or minimally overlapped with the processes from other cells; 4) the somata and dendritic processes were well-impregnated throughout the section thickness, with no apparent truncation of the dendritic arbor. Ten pyramidal neurons per region/brain were reconstructed with the aid of the Neurolucida software. Subsequently, somal area, total length of the dendritic tree, branching nodes and spine density (per 10 μm length) of the apical and basal dendrites, were obtained from a given selected neuron. 2.6. Statistical Analysis Imaging and numerical data for comparing groups were processed, with the mean ± SD calculated. Means were statistically analyzed by Student-t test or one-way ANOVA with posthoc Duncan’s multi-group comparisons when applicable. P < 0.05 was considered statistically significant. Figures were assembled with Photoshop 7.1, with brightness and contract adjusted as needed. 3. Results 3.1. LPS Injection Induced Immunoinflammatory Cellular/Molecular Changes To confirm the occurrence of chronic neuroinflammation, cerebral sections from the LPS and PBS groups were processed under identical conditions for the detection of immunoinflammatory proteins. Compared to PBS controls, increased immunoreactivity for MCH-II (Figures 1(A)-(E)), CD11b (not shown) and GFAP (not shown) occurred in the ipsilateral cortex and hippocampal formation in the LPS-injected brain sections. Specifically, MCH-II immunoreactive cells appeared to be largely glial cells (Figure 1(D) and Figure 1(E)). Double immunofluorescence showed that the majority of MCH-II labeled cells co-expressed the microglial marker CD11b (Figures 1(F)-(H)), although a few also colocalized with GFAP immunoreactivity, suggestive of a colocalization in astrocytes (Figures 1(I)-(K)). 3.2. LPS Injection Elevated BACE1 Protein Levels BACE1 protein levels were immunoblotted with a well-characterized rabbit antibody, which detects mature BACE1 protein migrating at ~70 kd [33] [34] [42] [43]. In the lysates of the ipsilateral hippocampi, immunob- X. H. Deng et al. 82 Figure 1. Representative images illustrating immunoinflammatory cellular/molecular changes in rat cortex and hippocampal formation following intracerebral lipolysaccharide (LPS) injection. Panel (A) shows minimal immunoreactivity of major histocompatibility complex class II molecules (MHC II) in the hippocampal CA1 to CA3 areas, the dentate gyrus (DG) and the overlaying cortex (Ctx) in a PBS-injected control rat, with the area of the dentate gyrus enlarged as (C). MHC II immunolabeling is increased in both the cortex and hippocampal formation in the LPS-injected animal (B), with the labeled cellular profiles appeared as glial cells at high magnifications ((D), (E)). Confocal double immunofluorescence shows a great extent of colocalization of MHC II reactivity among CD11b labeled microglial cells in the LPS-treated cerebrum ((F)-(H)). A small amount of MHC II labeled cells exhibit glial fibrillary acidic protein (GFAP) immunoreactivity, suggestive of a colocalization in activated astrocytes ((I)-(K)). Scale bar = 500 μm in A applying to B; equivalent to 250 μm for ((C), (D)); and to 75 μm for ((E)-(K)). lotted BACE1 signal was significantly increased relative to PBS-treated counterparts (Figure 2(A)). The mean optic density of BACE1 in the LPS group increased one fold (199.0% ± 52.8%) relative to control (100.0% ± 12.2%) (P = 0.043, t = 3.40, df = 3; two-tailed paired t-test) (Figure 2(A) and Figure 2(B)). Serving as an experimental as well as assay control [36]-[38], GFAP levels in the same cerebral samples were checked. In the ipsilateral hippocampal lysates, levels of GFAP were significantly (P = 0.027, t = 4.03, df = 3; two-tailed paired t-test) elevated in the LPS-treated (141.7% ± 11.4%) relative to PBS-treated (100% ± 13.4%) samples (Figure 2(A) and Figure 2(C)). 3.3. LPS Injection Induced Axonal Pathology with Enhanced Amyloidogenic Protein Expression In normal mammalian brains BACE1 immunoreactivity is largely expressed in the neuropil in a diffuse pattern X. H. Deng et al. 83 Figure 2. Western blot analysis showing elevation of the levels of β-secretase-1 (BACE1) and GFAP in the LPS-injected relative to PBS-treated (control) brains (n = 4/group), as assayed using hippocampal lysates ipsilateral to the injection. BACE1 levels in the LPS-treated group are about 2 times of that in the control ((A), (B)), while GFAP levels in the LPS group approach to 1.5 times of the control ((A), (C)), with the changes being significant for both proteins (paired two-tail t-tests). except for a distinct heavy labeling at selected neuronal terminal fields including the mossy fiber terminals and olfactory glomeruli. In human and transgenic animal brains with amyloid plaque pathology, BACE1 immunoreactivity is increased preferentially in swollen and sprouting axonal terminals or axonal dystrophic neurites [33]-[35] [42]. In the present study, BACE1 labeling in both cerebral hemispheres in the PBS injected brains and the contralateral cerebrum in the LPS-injected animals exhibited the aforementioned normal distribution pattern (Figure 3(A) and Figure 3(B)). On the contrary, BACE1 labeled neuritic structures emerged in the ipsilateral hemisphere of the LPS-treated brains in a site-specific manner. Thus, increased BACE labeling appeared in both the cortex and hippocampal formation especially evident around the needle track. At high magnification, these labeled profiles appeared as swollen sphericles and neuritic processes (Figures 3(B)-(F)). In the white matter, these neuritic processes appeared to spread from the needle track for a considerable distance (Figure 3(B) and Figure 3(D)). In the ipsilateral hippocampus, the BACE1 labeled swollen neurites occurred in the stratum pyramidale (s.p.) to stratum-lacunosum-moleculare (s.l.m.) of CA3 and the adjacent CA1 area (Figure 3(C)). A large amount of swollen sphericles extended along the stratum oriens (s.o.) of CA3 in the LPS-injected animals (Figure 3(E) and Figure 3(F)). At high magnifications, these BACE1 labeled neurites were mostly round or oval in shape, and varied in size. Some smaller sphericles were sometimes arranged in chains resembling enlarged axonal varicosities (Figure 3(C), Figure 3(D) and Figure 3(F)). In double immunofluorescence, the BACE1 immunoreactive swollen neurites exhibited a great extent of colocalization with APP (Figures 4(A)-(H)). In addition, the Aβ antibody 4G8 (which detects mouse APP, Aβ and likely APP β-C-terminal fragments, ref. 34) showed increased immunoreactivity in the same area with BACE1 labeled neurites (Figures 4(I)-(K)). A colocalization of BACE1 and 4G8 labeling was detected among some individual neuritic profiles. Notably, there existed a considerable amount of diffuse extracellular Aβ labeling (not colocalized with BACE1 reactivity), some of which appeared punctate (Figure 4(L)). The BACE1 labeled X. H. Deng et al. 84 Figure 3. Representative images illustrating the occurrence of BACE1 immunoreactive neuritic pathology after focal LPS injection in rat cerebrum. In the vehicle control (injection of phosphate-buffered saline, PBS), the pattern of BACE1 immunoreactivity is comparable between the ipsilateral and contralateral cortex and hippocampal formation, which is generally associated with neuropil except for a heavy labeling at the hippocampal mossy fiber (mf) terminals (A). In the LPS injected animal (B), BACE1 immunoreactivity is increased in the neuropil in the ipsilateral relative to contralateral cortex and hippocampal formation, especially apparent around the needle track (outline by arrows). At high magnification, swollen neurites are found in the cortex and hippocampal formation in the ipsilateral side, especially evident in the white matter, stratum radiatum (s.r.), stratum-lacunosum-moleculare (s.l.m.) and the striatum oriens (s.o.) that show little labeling in the normal conditions ((C), (D), (F)). Panels ((E), (F)) are taken from the section that is ~480 μm (12 × 30 μm) apart from (B), showing the distinct difference regarding the neuritic pathology between the two sides. The needle entry at the cortical surface is marked with an asterisk (*). Ctx: cortex (grey matter); DG: dentate gyrus; PC: parietal cortex; s.l.m.: stratum-lacunosum-moleculare; s.p.: stratum pyramidale. WM: white matter; Scale bar = 1 mm in (A) applying to (B); equivalent for 400 μm for ((C), (D)) and 200 μm for (E). sphericles and swollen processes were partially colocalized with the presynaptic marker synaptophysin (Figures 4(M)-(P)). In contrast, the BACE1 labeled swollen neurites did not colocalize with MAP2, which is clearly expressed in dendrites and somata of nearby pyramidal neurons (Figures 4(R)-(U)). 3.4. LPS Injection Induced Somotodendritic Changes in Cerebral Principal Neurons In our Golgi preparations, pyramidal neurons around layer III were consistently impregnated in all brain samples (Figure 5(A)). In the hippocampal proper, the curving area of CA3, whereby the s.p. continues dorsally from the X. H. Deng et al. 85 Figure 4. Confocal double immunofluorescent images showing LPS induced axonal pathology associated with increased labeling of amyloidogenic proteins. All images are taken from the CA3 area of the hippocampus ipsilateral to the intracerebral injection of PBS ((A)-(C)) or LPS ((E)-(U)). Antibody markers and color channels are as indicated. Panels ((A)-(C)) show double labeling of BACE1 and β-amyloid precursor protein (APP) in ipsilateral CA3 of the control animal, with the former expressed predominantly in the mossy fiber (mf) terminals, and the latter largely in somata of CA3 pyramidal neurons. Note that no abnormal neurites are present in the strum oriens (s.o.). Panels ((E)-(H)) illustrate the occurrence of BACE1/APP double labeled swollen neurites (examples are indicated by arrows) in the s.o. of the ipsilateral hippocampus of the LPS injected rat. Panels (I-L) show that these BACE1 positive neurites are locally associated with increased 4G8 labeling within (pointed by arrows) as well as outside (arrowheads) the swollen terminals in the cortex and CA1. Panels ((M)-(P)) show that a partial coexpression of synaptophysin (SYN) among the BACE1 labeled swollen neurites in the s.o. of the LPS injected ipsilateral hippocampus, which appear in yellow in the merged image (arrows, P). Panels (R-U) show that there is no colocalization of the microtubule associated protein-2 (MAP2) in the BACE1 labeled swollen neurites. MAP2 labeling is distinctly associated with the somata and dendrites of pyramidal neurons in the stratum pyramidale (s.p.). DAPI counterstain is included in panels (L) and (U), showing that the BACE1 labeled elements are not somatic. Scale bar = 200 μm in (A) applying to ((B), (C), (E)-(G) and (R)-(T)), equivalent to 100 μm for (I)-(K), (M)-(O)), 50 μm for ((D), (H), (U)) and 25 μm for ((L), (P)). CA1 direction and turns ventrally toward the dentate gyrus, could be defined systematically across brains. Therefore, we decided to use Golgi-stained pyramidal neurons in cortical layer III and around the dorsoventral turning area of CA3 for automated Neurolucida reconstruction and morphometry. Measurements from reconstructed layer III and CA1 pyramidal neurons in the ipsilateral hemisphere of the LPS and PBS treated brains X. H. Deng et al. 86 Figure 5. Quantitative Golgi study of somal and dendritic changes in cortical layer III pyramidal neurons in LPS-injected relative to PBS-injected rats. Panel (A) shows an example of Golgi stain in the parietal neocortex (ipsilateral to PBS injection), note that layer III pyramidal neurons are relatively well-labeled. Panels ((B), (C)) show representative Neurolucida reconstructions of impregnated layer III pyramidal neurons from a LPS (B) and a PBS (C) injected brains, with their apical and basal dendrites and dendritic branching points illustrated in color. Bar graphs ((D)-(G)) summarize the parameters obtained from 10 constructed neurons in each brain (n = 4/group). The means of somal perimeter and somal area show a trend of reduction in the LPS relative to PBS (control) groups, although the difference is not statistically significant (E). The means of total dendritic length tends to reduce in the LPS group relative to control, with significant difference for that of the basal dendritic tree (F). The branching nodes (points) are reduced in the LPS group on the basal dendritic tree (G). Overall spine density is significantly reduced on the dendritic tree of Golgi-impregnated cortical pyramidal neurons in the LPS group relative to control (H). Scale bar = 200 μm in (A). (10 cells per region per brain) were compared quantitatively (Figures 5(A)-(C) and Figures 6(A)-(D)). The somal perimeters of the layer III pyramidal neurons were comparable between the LPS (58.6 ± 7.4 µm, mean ± S.D., same format below) and PBS (61.7 ± 9.9 µm) groups (P > 0.05, two-tail paired t-test; same statistical test below) (Figure 5(D)). The mean somal area of layer III pyramidal neurons was reduced in the LPS (168.4 ± 21.6 µm2 ) relative to the PBS (216.5 ± 45.3 µm2 ) groups (P < 0.001). The total length of the dendritic processes of the layer III pyramidal neurons tended to be reduced in the LPS relative to PBS groups (Figure 5(E)). Thus, the total length of the apical dendrites was 647.4 ± 182.9 µm in the LPS group as compared to 834.9 ± 228.8 µm in the PBS group, with the means not statistically different (P > 0.05). The means of the total length of the basal dendrites were 711.4 ± 226.9 µm and 1122.0 ± 320.7 µm in the LPS and PBS groups, respectively, showing a significant difference (P < 0.001). The means of the branching points (nodes) on the apical dendrites were 7.0 ± 2.5 and 8.5 ± 2.4 in the LPS and the PBS groups, respectively, while they were 7.2 ± 1.3 X. H. Deng et al. 87 Figure 6. Quantitative Golgi study of somal and dendritic changes in in CA3 pyramidal neurons in LPS-injected relative to PBS-injected rat brains. Panel (A) shows an example of Golgi-stained CA3 pyramidal neurons located around dorsal to ventral turning area of the stratum pyramidale (s.r.). Basal dendrites in the stratum oriens (s.o.) and apical dendrites in the stratum radiatum (s.r.) are visible, with dendritic spines clearly labeled at high resolution (B). Panels ((C), (D)) show examples of Neurolucida reconstructions of CA3 pyramidal neurons from LPS (C) and a PBS (D) injected brains. The reduction in mean somal area (SA) in the LPS relative to PBS groups is statistically significant, whereas the reduction in the somal perimeter (SP) is not (E). The reduction in the total dendritic length in the LPS relative to PBS groups is significant for both the basal and apical trees (F). Scale bar = 50 μm in (A) and 5 μm for (B). and 10.6 ± 2.7 for the two groups on the basal dendrites. Statistical tests reported a significant difference between the two groups for the means of the basal dendritic but not the apical measurements (Figure 5(F)). The number of spines per unit length of dendrite was reduced significantly on the layer III pyramidal neurons in the LPS relative to PBS groups (Figure 5(G)). Data from the apical and basal dendrites combined, showed spine density to be 6.3 ± 0.9 vs. 8.0 ± 1.1 for the LPS relative to PBS groups (P < 0.0001). For the reconstructed CA3 pyramidal neurons (Figures 6(A)-(D)), both the somal perimeter and somal area of the pyramidal neurons tended to reduce in the LPS relative to PBS groups (Figure 6(E)). Thus, the mean somal perimeters were 47.8 ± 11.0 µm (mean ± S.D., same format below) and 67.8 ± 12.3 µm in the LPS and PBS treated animals, respectively (P > 0.05, two-tail paired t-test, same statistical test below). The reduction in the somal area of CA3 pyramidal neurons was significant (P < 0.001) in the LPS group (144.8 ± 40.4 µm2 ) compared to the PBS group (278.3 ± 60.2 µm2 ). There were significant decreases in the total lengths of the apical dendrites (320.0 ± 52.6 vs. 690.0 ± 63.6 µm; P < 0.005) and the basal dendrites (534.6 ± 70.6 vs. 967.0 ± 90.6 µm; P < 0.001) in the LPS compared to PBS groups (Figure 6(F)). The number of branching points on the apical dendrites (4.3 ± 2.1 vs. 8.3 ± 2.1; P < 0.001) and basal dendrites (3.9 ± 1.5 vs 7.2 ± 1.7; P < 0.001) of CA1 pyramidal neurons was significantly reduced in the LPS relative to PBS groups (Figure 6(G)). Finally, the density of spines on the apical and basal dendrites of the CA3 pyramidal neurons was significantly reduced (P < 0.001) in the LPS animals (10.5 ± 3.2) relative to PBS controls (18.0 ± 3.3) (Figure 6(H)). 4. Discussion 4.1. The LPS Model for Studying Chronic Neurodegeneration and Alzheimer’s Disease The LPS model is increasingly used in mechanistic and translational studies into chronic neurodegenerative dis X. H. Deng et al. 88 eases such as AD and PD [15] [16] [38] [44]-[50]. This relatively simple experimental approach offers yet a number of advantages in elucidating the neurobiology of aging and age-relative diseases. This model recapitulates many cellular/molecular events of neuroinflammation, which is considered an early, perhaps fundamental, causal factor in chronic neuronal and synaptic degeneration [8]-[14] [36]-[41]. The model also reproduces some defining clinical manifestations of human neurodegenerative diseases in rodents [44]-[46]. For instance, peripheral and central LPS administration induces learning/memory deficits in experimental animals analogous to AD-like cognitive decline [39]-[41]. Importantly, LPS treatment elicits neuropathological and behavioral changes in wild-type animals [47]-[50]. As neurodegenerative diseases occur mostly in a sporadic nature, the LPS model serves an ideal system to address pathogenic mechanisms underlying common human neurological disorders. Specifically, this study examined whether inflammation is a consequence or cause of dystrophic amyloidogenic neuritis. We have shown in a recent study that unilateral intracerebral injection of ~10 μg LPS is sufficient to induce spatial learning and memory impairments in adult rats. Upregulation of immunoinflammatory molecules including PirB and GFAP is evident in the LPS-treated brains, occurring largely in the ipsilateral cortex and hippocampal formation [41]. In the present study we observe that this same dose of LPS injection dramatically induces MHC II expression in the ipsilateral neocortex and hippocampal formation. This modulation appears to be largely associated with microglial activation, a key finding in the initial studies that establish this model of neurological diseases [36] [39]. Consistent with other reports [51], LPS-induced MHC II upregulation also appears to occur to a lesser extent in activated astrocytes that express high levels of GFAP. Thus, our data are consistent with the notion that intracranial LPS injection induces profound glial immunoinflammatory responses in the brain. 4.2. Axonal and Dendritic Pathology in LPS-Induced Neuroinflammation Besides glial responses, neuronal and synaptic alterations have been reported in rodent LPS models anatomically and electrophysiologically. Local LPS infusion causes increased cell/neuronal death in the entorhinal cortex [52] and substantia nigra [53] in rats as well as the hippocampus in ICR mice [54]. In BALB/c mice, the length and branching points of apical dendrites of CA1 pyramidal neurons are reduced in old but not young adults 72 hours following intraperitoneal LPS injection, while no change in spine density is found on these neurons [55]. In C57BL/6 mice, an increase in the density of thin spines on the dendrites of adult-born granule cells is found in the inner but not outer molecular layer 28 days following intrahippocampal LPS injection [56]. In adult rats, intrahippocampal LPS injection does not alter the intrinsic membrane properties, dendritic arborization or spine formation of the newly generated granule cells, but affect the overall network activity in the hippocampal neural circuitries [57]. LPS treatment can alter hippocampal synaptoplasticity in rats as measured by long-term potential and long-term depression [52] [58]-[60]. Here we demonstrate pre- and post-synaptic alterations among cortical and hippocampal neurons in LPS relative to PBS injected rats. Microscopically evident axonal pathology is featured by aberrant sprouting and swelling, and increased expression of APP, BACE1 and synaptophysin, but not MAP2. These abnormal axonal profiles seen following LPS treatment resemble the dystrophic neurites seen in models of injury that exhibit an increase in APP or A-beta [61] [62], although they do not arrange in typical rosette-like clusters as seen around established compact plaques [32]. Our Golgi data indicate that focal LPS injection causes a significant decrease in dendritic length and nodes on the basal tree as well as a reduction of spine density on the entire dendritic tree of layer III cortical principal neurons. The impact of focally injected LPS appears to be greater on CA3 pyramidale neurons, including significant reductions of somal size, lengths of basal and apical dendrites, branching points of the basal and apical dendrites, and overall spine density. The vulnerability of CA3 pyramidale neurons to LPS toxicity might relate to an intrinsic property of these cells in regard to synaptoplasticity. The dendrites and spines of CA3 pyramidal neurons belong to one of most plastic network systems in the brain—they receive major presynaptic inputs from the mossy fiber terminals that are constantly renewed in the process of adult neurogenesis of the dentate granule cells [63]. Alternatively, the observed variable somotodendritic changes between the cortical and hippocampal pyramidal neurons may reflect a potential spatial or dose-related effect by the injected toxin (i.e., the proximity to the injection site), which can be expected since the inflammation and BACE1 upregulation are not evident in the contralateral hemisphere. X. H. Deng et al. 89 4.3. Amyloid Pathogenesis in LPS-Induced Neuroinflammation LPS administration has been shown to influence amyloid pathogenesis via mixed cellular modulations in wild-type and transgenic rodents. Chronic intraventricular LPS infusion in rats induces intraneuronal Aβ immunoreactivity in the hippocampus [37]. Repeated intraperitoneal LPS injections in ICR mice result in increased intraneuronal and extracellular Aβ42 immunoreactivity in the cortex and hippocampus, with biochemical data indicating reduced α-secretase activity, increased γ-secretase activity, and elevated BACE1 protein and activity in tissue lysate [64]. In transgenic models of AD, LPS administration is shown to increase APP and Aβ immunoreactivity in neuronal somata [65], elevate γ-secretase activity [66] and accelerate plaque deposition [67]. However, other reports show reduced amyloid deposition via enhanced Aβ clearance by activated microglia after LPS administration [68] [69]. Being the obligatory enzyme for Aβ genesis, BACE1 is at the crossing point potentially linking cellular and signaling substrates to amyloid pathogenesis. BACE1 elevation is associated with axonal pathology during plaque development in the brains of transgenic AD models as well as aged and AD human subjects [32] [70]. As elaborated in the preceding section, we have identified axonal sprouting and swelling associated with enhanced APP, BACE1 and Aβ antibody reactivity in the ipsilateral cortex and hippocampal formation in LPS treated rat brains, especially evident around the needle track. This finding clearly indicates that chronic neuroinflammation can act as an inductive factor for amyloidogenic axonal pathology in wild-type adult mammalian brain. More basic questions can be asked as to why axonal sprouting/dystrophy, dendritic/spine degeneration and glial activation develop in partnership under neuroinflammatory conditions. One possibility is that the toxin interrupts axonal transport causing accumulation of the amyloidogenic proteins and other neuronal molecules [31]. This is consistent with the observation that the presynaptic marker synaptophysin is present only among a subset of the BACE1 immunoreactive swollen neurites. One may also speculate that this pathogenic correlation potentially involves the triadic organization of the synapse—presynaptic and postsynaptic terminals engulfed by glial processes. Neuroinflammation could initially induce some disengagement between the synaptic terminals and glial scaffold. For instance, inflammation-associated structural and functional changes in the glial cells may disrupt the fitness, integrity and plasticity of the pre- and post-synaptic terminals. This may eventually lead to major degenerative changes in the postsynaptic components, resulting in dendritic shrinkage and spine loss. The presynaptic components may also largely regress in the course of synaptic degeneration. However, some axon and presynaptic terminals might otherwise undergo an aggressive regenerative attempt because of the loss of appropriate postsynaptic targets. In this sense, the aberrant sprouting and dystrophy of the axonal terminals may be considered as a form of maladaptive neuronal regeneration, with the activated amyloidogenic machinery being a part of the its complex molecular dyshomeostasis. In summary, the present study demonstrates that chronic neuroinflammation induced by intracerebral LPS injection promotes the amyloidogenic cascade in dystrophic neurites by upregulation of BACE1 and APP in the brain. This modulation is evident in axon terminals that exhibit dystrophic-like morphology. LPS injection also induces degenerative changes in postsynaptic components manifested as dendritic shrinkage and spine loss among cortical and hippocampal pyramidal neurons. Together with the immunoinflammatory responses of the glial cells, the LPS model recapitulates multiple cellular and molecular deficits seen in aging and AD brains. The synaptic triad could be the initial site as well as center of neural degeneration and aberrant regeneration, with its progression ultimately leading to overt neuritic amyloid pathology and cognitive deficits. Acknowledgements This study was supported by the National Natural Science Foundation of China (#81171091, #81171160), Natural Science Foundation of Hunan Province (13JJ5016), Central South University (CSUZC2014035), Center for Alzheimer’s Disease and Related Disorders (P.R.P), and the Intramural research program of National Institute on Aging, NIH (Z01-IAAG000944-04 to H.C.). References [1] McGeer, E.G. and McGeer, P.L. (2010) Neuroinflammation in Alzheimer’s Disease and Mild Cognitive Impairment: A Field in Its Infancy. Journal of Alzheimer’s Disease, 19, 355-361. [2] Qian, L., Flood, P.M. and Hong, J.S. (2010) Neuroinflammation is a Key Player in Parkinson’s Disease and a Prime X. H. Deng et al. 90 Target for Therapy. Journal of Neural Transmission, 117, 971-979. http://dx.doi.org/10.1007/s00702-010-0428-1 [3] Broussard, G.J., Mytar, J., Li, R.C. and Klapstein, G.J. (2012) The Role of Inflammatory Processes in Alzheimer’s Disease. Inflammopharmacology, 20, 109-126. [4] Luessi, F., Siffrin, V. and Zipp, F. (2012) Neurodegeneration in Multiple Sclerosis: Novel Treatment Strategies. Expert Review of Neurotherapeutics, 12, 1061-1076. http://dx.doi.org/10.1586/ern.12.59 [5] Rao, J.S., Kellom, M., Kim, H.W., Rapoport, S.I. and Reese, E.A. (2012) Neuroinflammation and Synaptic Loss. Neurochemical Research, 37, 903-910. http://dx.doi.org/10.1007/s11064-012-0708-2 [6] Blandini, F. (2013) Neural and Immune Mechanisms in the Pathogenesis of Parkinson’s Disease. Journal of NeuroImmune Pharmacology, 8, 189-201. http://dx.doi.org/10.1007/s11481-013-9435-y [7] Breunig, J.J., Guillot-Sestier, M.V. and Town, T. (2013) Brain Injury, Neuroinflammation and Alzheimer’s Disease. Frontiers in Aging Neuroscience, 11, 26. [8] Enciu, A.M. and Popescu, B.O. (2013) Is There a Causal Link between Inflammation and Dementia? BioMed Research International, 2013, 316495. http://dx.doi.org/10.1155/2013/316495 [9] Ownby, R.L. (2010) Neuroinflammation and Cognitive Aging. Current Psychiatry Reports, 12, 39-45. http://dx.doi.org/10.1007/s11920-009-0082-1 [10] Pizza, V., Agresta, A., D’Acunto, C.W., Festa, M. and Capasso, A. (2011) Neuroinflammation and Ageing: Current Theories and an Overview of the Data. Reviews on Recent Clinical Trials, 6, 189-203. http://dx.doi.org/10.2174/157488711796575577 [11] Cagnin, A., Brooks, D.J., Kennedy, A.M., Gunn, R.N., Myers, R. and Turkheimer, F.E. (2001) In-Vivo Measurement of Activated Microglia in Dementia. Lancet, 358, 461-467. http://dx.doi.org/10.1016/S0140-6736(01)05625-2 [12] Butcher, S.K. and Lord, J.M. (2004) Stress Responses and Innate Immunity: Aging as a Contributory Factor. Aging Cell, 3, 151-160. http://dx.doi.org/10.1111/j.1474-9728.2004.00103.x [13] Eikelenboom, P., van Exel, E., Hoozemans, J.J., Veerhuis, R., Rozemuller, A.J. and van Gool, W.A. (2010) Neuroinflammation—An Early Event in Both the History and Pathogenesis of Alzheimer’s Disease. Neurodegenerative Diseases, 7, 38-41. http://dx.doi.org/10.1159/000283480 [14] Wang, X., Wang, W., Li, L., Perry, G., Lee, H.G. and Zhu, X. (2013) Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochimica et Biophysica Acta, S0925-4439(13)00323-2. [15] Katafuchi, T., Ifuku, M., Mawatari, S., Noda, M., Miake, K., Sugiyama, M. and Fujino, T. (2012) Effects of Plasmalogens on Systemic Lipopolysaccharide-Induced Glial Activation and β-Amyloid Accumulation in Adult Mice. Annals of the New York Academy of Sciences, 1262, 85-92. http://dx.doi.org/10.1111/j.1749-6632.2012.06641.x [16] Krstic, D., Madhusudan, A., Doehner, J., Vogel, P., Notter, T., Imhof, C., Manalastas, A., Hilfiker, M., Pfister, S., Schwerdel, C., Riether, C., Meyer, U. and Knuesel, I. (2012) Systemic Immune Challenges Trigger and Drive AlzheimerLike Neuropathology in Mice. Journal of Neuroinflammation, 9, 151. http://dx.doi.org/10.1186/1742-2094-9-151 [17] Kraft, A.W., Hu, X., Yoon, H., Yan, P., Xiao, Q., Wan, Y., Gil, S.C., Brown, J., Wilhelmsson, U., Restivo, J.L., Cirrito, J.R., Holtzman, D.M., Kim, J., Pekny, M. and Lee, J.M. (2013) Attenuating Astrocyte Activation Accelerates Plaque Pathogenesis in APP/PS1 Mice. FASEB Journal, 27, 187-198. http://dx.doi.org/10.1096/fj.12-208660 [18] Medeiros, R. and LaFerla, F.M. (2013) Astrocytes: Conductors of the Alzheimer Disease Neuroinflammatory Symphony. Experimental Neurology, 239, 133-138. http://dx.doi.org/10.1016/j.expneurol.2012.10.007 [19] Zhao, J., O’Connor, T. and Vassar, R. (2011) The Contribution of Activated Astrocytes to Aβ Production: Implications for Alzheimer’s Disease Pathogenesis. Journal of Neuroinflammation, 8, 150. http://dx.doi.org/10.1186/1742-2094-8-150 [20] Sondag, C.M., Dhawan, G. and Combs, C.K. (2009) Beta Amyloid Oligomers and Fibrils Stimulate Differential Activation of Primary Microglia. Journal of Neuroinflammation, 6, 1. http://dx.doi.org/10.1186/1742-2094-6-1 [21] Maezawa, I., Zimin, P.I., Wulff, H. and Jin, L.W. (2011) Amyloid-Beta Protein Oligomer at Low Nanomolar Concentrations Activates Microglia and Induces Microglial Neurotoxicity. Journal of Biological Chemistry, 286, 3693-3706. http://dx.doi.org/10.1074/jbc.M110.135244 [22] DeKosky, S.T. and Scheff, S.W. (1990) Synapse Loss in Frontal Cortex Biopsies in Alzheimer’s Disease: Correlation with Cognitive Severity. Annals of Neurology, 27, 457-464. http://dx.doi.org/10.1002/ana.410270502 [23] Terry, R.D., Masliah, E., Salmon, D.P., Butters, N., Deteresa, R., Hill, R., Hansen, L.A. and Katzman, R. (1991) Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Annals of Neurology, 30, 572-580. http://dx.doi.org/10.1002/ana.410300410 [24] Masliah, E., Mallory, M., Alford, M., DeTeresa, R., Hansen, L.A., McKeel Jr., D.W. and Morris, J.C. (2001) Altered Expression of Synaptic Proteins Occurs Early during Progression of Alzheimer’s Disease. Neurology, 56, 127-129. http://dx.doi.org/10.1212/WNL.56.1.127 X. H. Deng et al. 91 [25] Heinonen, O., Soininen, H., Sorvari, H., Kosunen, O., Paljarvi, L., Koivisto, E. and Riekkinen, P.J. (1995) Loss of Synaptophysin-Like Immunoreactivity in the Hippocampal Formation Is an Early Phenomenon in Alzheimer’s Disease. Neuroscience, 64, 375-384. http://dx.doi.org/10.1016/0306-4522(94)00422-2 [26] Reddy, P.H., Mani, G., Park, B.S., Jacques, J., Murdoch, G., Whetsell Jr., W., Kaye, J. and Manczak, M. (2005) Differential Loss of Synaptic Proteins in Alzheimer’s Disease: Implications for Synaptic Dysfunction. Journal of Alzheimer’s Disease, 7, 103-117. [27] Scheff, S.W., Price, D.A., Schmitt, F.A., Scheff, M.A. and Mufson, E.J. (2011) Synaptic Loss in the Inferior Temporal Gyrus in Mild Cognitive Impairment and Alzheimer’s Disease. Journal of Alzheimer’s Disease, 24, 547-557. [28] D’Amelio, M. and Rossini, P.M. (2012) Brain Excitability and Connectivity of Neuronal Assemblies in Alzheimer’s Disease: From Animal Models to Human Findings. Progress in Neurobiology, 99, 42-60. http://dx.doi.org/10.1016/j.pneurobio.2012.07.001 [29] Berchtold, N.C., Coleman, P.D., Cribbs, D.H., Rogers, J., Gillen, D.L. and Cotman, C.W. (2013) Synaptic Genes Are Extensively Downregulated across Multiple Brain Regions in Normal Human Aging and Alzheimer’s Disease. Neurobiology of Aging, 34, 1653-1661. http://dx.doi.org/10.1016/j.neurobiolaging.2012.11.024 [30] Knobloch, M. and Mansuy, I.M. (2008) Dendritic Spine Loss and Synaptic Alterations in Alzheimer’s Disease. Molecular Neurobiology, 37, 73-82. http://dx.doi.org/10.1007/s12035-008-8018-z [31] Kanaan, N.M., Pigino, G.F., Brady, S.T., Lazarov, O., Binder, L.I. and Morfini, G.A. (2013) Axonal Degeneration in Alzheimer’s Disease: When Signaling Abnormalities Meet the Axonal Transport System. Experimental Neurology, 246, 44-53. http://dx.doi.org/10.1016/j.expneurol.2012.06.003 [32] Yan, X.X., Ma, C., Gai, W.P., Cai, H. and Luo, X.G. (2014) Can BACE1 Inhibition Mitigate Early Axonal Pathology in Neurological Diseases? Journal of Alzheimer’s Disease, 38, 705-718. [33] Zhang, X.M., Cai, Y., Xiong, K., Cai, H., Luo, X.G., Feng, J.C., Clough, R.W., Struble, R.G., Patrylo, P.R. and Yan, X.X. (2009) β-Secretase-1 Elevation in Transgenic Mouse Models of Alzheimer’s Disease Is Associated with Synaptic/ Axonal Pathology and Amyloidogenesis: Implications for Neuritic Plaque Development. European Journal of Neuroscience, 30, 2271-2283. http://dx.doi.org/10.1111/j.1460-9568.2009.07017.x [34] Cai, Y., Zhang, X.M., Macklin, L.N., Cai, H., Luo, X.G., Oddo, S., Laferla, F.M., Struble, R.G., Rose, G.M., Patrylo, P.R. and Yan, X.X. (2012) BACE1 Elevation Is Involved in Amyloid Plaque Development in the Triple Transgenic Model of Alzheimer’s Disease: Differential Aβ Antibody Labeling of Early-Onset Axon Terminal Pathology. Neurotoxicity Research, 21, 160-174. http://dx.doi.org/10.1007/s12640-011-9256-9 [35] Li, J.M., Xue, Z.Q., Deng, S.H., Luo, X.G., Patrylo, P.R., Rose, G.W., Cai, H., Cai, Y. and Yan, X.X. (2013) Amyloid Plaque Pathogenesis in 5XFAD Mouse Spinal Cord: Retrograde Transneuronal Modulation after Peripheral Nerve Injury. Neurotoxicity Research, 24, 1-14. http://dx.doi.org/10.1007/s12640-012-9355-2 [36] Xu, J. and Ling, E.A. (1994) Upregulation and Induction of Surface Antigens with Special Reference to MHC Class II Expression in Microglia in Postnatal Rat Brain Following Intravenous or Intraperitoneal Injections of Lipopolysaccharide. Journal of Anatomy, 184, 285-296. [37] Hauss-Wegrzyniak, B., Dobrzanski, P., Stoerh, J.D. and Wenk, G.L. (1998) Chronic Neuroinflammation in Rats Reproduces Components of the Neurobiology of Alzheimer’s Disease. Brain Research, 780, 294-303. http://dx.doi.org/10.1016/S0006-8993(97)01215-8 [38] Sugaya, K., Chou, S., Xu, S.J. and McKinney, M. (1998) Indicators of Glial Activation and Brain Oxidative Stress after Intraventricular Infusion of Endotoxin. Molecular Brain Research, 58, 1-9. http://dx.doi.org/10.1016/S0169-328X(97)00365-3 [39] Tanaka, S., Ide, M., Shibutani, T., Ohtaki, H., Numazawa, S., Shioda, S. and Yoshida, T. (2006) LipopolysaccharideInduced Microglial Activation Induces Learning and Memory Deficits without Neuronal Cell Death in Rats. Journal of Neuroscience Research, 83, 557-566. http://dx.doi.org/10.1002/jnr.20752 [40] Lee, J.W., Lee, Y.K., Yuk, D.Y., Choi, D.Y., Ban, S.B., Oh, K.W. and Hong, J.T. (2008) Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment through Enhancement of Beta-Amyloid Generation. Journal of Neuroinflammation, 5, 37. http://dx.doi.org/10.1186/1742-2094-5-37 [41] Deng, X.H., Ai, W.M., Lei, D.L., Luo, X.G., Yan, X.X. and Li, Z. (2012) Lipopolysaccharide Induces Paired Immunoglobulin-Like Receptor B (PirB) Expression, Synaptic Alteration, and Learning-Memory Deficit in Rats. Neuroscience, 209, 161-170. http://dx.doi.org/10.1016/j.neuroscience.2012.02.022 [42] Laird, F.M., Cai, H., Savonenko, A.V., Farah, M.H., He, K., Melnikova, T., Wen, H., Chiang, H.C., Xu, G., Koliatsos, V.E., Borchelt, D.R., Price, D.L., Lee, H.K. and Wong, P.C. (2005) BACE1, a Major Determinant of Selective Vulnerability of the Brain to Amyloid-β Amyloidogenesis, Is Essential for Cognitive, Emotional, and Synaptic Functions. Journal of Neuroscience, 25, 11693-11709. http://dx.doi.org/10.1523/JNEUROSCI.2766-05.2005 X. H. Deng et al. 92 [43] Xiong, K., Cai, H., Luo, X.G., Struble, R.G., Clough, R.W. and Yan, X.X. (2007) Mitochondrial Respiratory Inhibition and Oxidative Stress Elevate β-Secretase (BACE1) Proteins and Activity in Vivo in the Rat Retina. Experimental Brain Research, 181, 435-446. http://dx.doi.org/10.1007/s00221-007-0943-y [44] Dutta, G., Zhang, P. and Liu, B. (2008) The Lipopolysaccharide Parkinson’s Disease Animal Model: Mechanistic Studies and Drug Discovery. Fundamental & Clinical Pharmacology, 22, 453-464. http://dx.doi.org/10.1111/j.1472-8206.2008.00616.x [45] Lee, D.C., Rizer, J., Selenica, M.L., Reid, P., Kraft, C., Johnson, A., Blair, L., Gordon, M.N., Dickey, C.A. and Morgan, D. (2010) LPS-Induced Inflammation Exacerbates Phospho-Tau Pathology in rTg4510 Mice. Journal of Neuroinflammation, 7, 56. http://dx.doi.org/10.1186/1742-2094-7-56 [46] Hoban, D.B., Connaughton, E., Connaughton, C., Hogan, G., Thornton, C., Mulcahy, P., Moloney, T.C. and Dowd, E. (2013) Further Characterisation of the LPS Model of Parkinson’s Disease: A Comparison of Intra-Nigral and IntraStriatal Lipopolysaccharide Administration on Motor Function, Microgliosis and Nigrostriatal Neurodegeneration in the Rat. Brain, Behavior, and Immunity, 27, 91-100. http://dx.doi.org/10.1016/j.bbi.2012.10.001 [47] Ifuku, M., Katafuchi, T., Mawatari, S., Noda, M., Miake, K., Sugiyama, M. and Fujino, T. (2012) Anti-Inflammatory/ Anti-Amyloidogenic Effects of Plasmalogens in Lipopolysaccharide-Induced Neuroinflammation in Adult Mice. Journal of Neuroinflammation, 9, 197. http://dx.doi.org/10.1186/1742-2094-9-197 [48] Martin, S.A., Pence, B.D., Greene, R.M., Johnson, S.J., Dantzer, R., Kelley, K.W. and Woods, J.A. (2013) Effects of Voluntary Wheel Running on LPS-Induced Sickness Behavior in Aged Mice. Brain, Behavior, and Immunity, 29, 113- 123. http://dx.doi.org/10.1016/j.bbi.2012.12.014 [49] Samanani, S., Mishra, M., Silva, C., Verhaeghe, B., Wang, J., Tong, J. and Yong, V.W. (2013) Screening for Inhibitors of Microglia to Reduce Neuroinflammation. CNS & Neurological Disorders-Drug Targets, 12, 741-749. http://dx.doi.org/10.2174/18715273113126660177 [50] Asti, A. and Gioglio, L. (2014) Can a Bacterial Endotoxin Be a Key Factor in the Kinetics of Amyloid Fibril Formation? Journal of Alzheimer’s Disease, 39, 169-179. [51] Morga, E., Faber, C. and Heuschling, P. (1999) Regional Heterogeneity of the Astroglial Immunoreactive Phenotype: Effect of Lipopolysaccharide. Journal of Neuroscience Research, 57, 941-952. http://dx.doi.org/10.1002/(SICI)1097-4547(19990915)57:6<941::AID-JNR20>3.0.CO;2-Z [52] Hauss-Wegrzyniak, B., Lynch, M.A., Vraniak, P.D. and Wenk, G.L. (2002) Chronic Brain Inflammation Results in Cell Loss in the Entorhinal Cortex and Impaired LTP in Perforant Path-Granule Cell Synapses. Experimental Neurology, 176, 336-341. http://dx.doi.org/10.1006/exnr.2002.7966 [53] Burguillos, M.A., Hajji, N., Englund, E., Persson, A., Cenci, A.M., Machado, A., Cano, J., Joseph, B. and Venero, J.L. (2011) Apoptosis-Inducing Factor Mediates Dopaminergic Cell Death in Response to LPS-Induced Inflammatory Stimulus: Evidence in Parkinson’s Disease Patients. Neurobiology of Disease, 41, 177-188. http://dx.doi.org/10.1016/j.nbd.2010.09.005 [54] Lee, J.W., Lee, Y.K., Yuk, D.Y., Choi, D.Y., Ban, S.B., Oh, K.W. and Hong, J.T. (2008) Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment through Enhancement of Beta-Amyloid Generation. Journal of Neuroinflammation, 5, 37. http://dx.doi.org/10.1186/1742-2094-5-37 [55] Richwine, A.F., Parkin, A.O., Buchanan, J.B., Chen, J., Markham, J.A., Juraska, J.M. and Johnson, R.W. (2008) Architectural Changes to CA1 Pyramidal Neurons in Adult and Aged Mice after Peripheral Immune Stimulation. Psychoneuroendocrinology, 33, 1369-1377. http://dx.doi.org/10.1016/j.psyneuen.2008.08.003 [56] Chugh, D., Nilsson, P., Afjei, S.A., Bakochi, A. and Ekdahl, C.T. (2013) Brain Inflammation Induces Post-Synaptic Changes during Early Synapse Formation in Adult-Born Hippocampal Neurons. Experimental Neurology, 250, 176- 188. http://dx.doi.org/10.1016/j.expneurol.2013.09.005 [57] Jakubs, K., Bonde, S., Iosif, R.E., Ekdahl, C.T., Kokaia, Z., Kokaia, M. and Lindvall, O. (2008) Inflammation Regulates Functional Integration of Neurons Born in Adult Brain. Journal of Neuroscience, 28, 12477-12488. http://dx.doi.org/10.1523/JNEUROSCI.3240-08.2008 [58] Commins, S., O’Neill, L.A. and O’Mara, S.M. (2001) The Effects of the Bacterial Endotoxin Lipopolysaccharide on Synaptic Transmission and Plasticity in the CA1-Subiculum Pathway in Vivo. Neuroscience, 102, 273-280. http://dx.doi.org/10.1016/S0306-4522(00)00498-X [59] Jo, J.H., Park, E.J., Lee, J.K., Jung, M.W. and Lee, C.J. (2001) Lipopolysaccharide Inhibits Induction of Long-Term Potentiation and Depression in the Rat Hippocampal CA1 Area. European Journal of Pharmacology, 422, 69-76. http://dx.doi.org/10.1016/S0014-2999(01)01075-5 [60] Min, S.S., Quan, H.Y., Ma, J., Lee, K.H., Back, S.K., Na, H.S., Han, S.H., Yee, J.Y., Kim, C., Han, J.S. and Seol, G.H. (2009) Impairment of Long-Term Depression Induced by Chronic Brain Inflammation in Rats. Biochemical and Biophysical Research Communications, 383, 93-97. http://dx.doi.org/10.1016/j.bbrc.2009.03.133 X. H. Deng et al. 93 [61] Gentleman, S.M., Nash, M.J., Sweeting, C.J., Graham, D.I. and Roberts, G.W. (1993) β-Amyloid Precursor Protein (βAPP) as a Marker for Axonal Injury after Head Injury. Neuroscience Letters, 160, 139-144. http://dx.doi.org/10.1016/0304-3940(93)90398-5 [62] Chen, X.H., Johnson, V.E., Uryu, K., Trojanowski, J.Q. and Smith, D.H. (2009) A Lack of Amyloid Beta Plaques Despite Persistent Accumulation of Amyloid β in Axons of Long-Term Survivors of Traumatic Brain Injury. Brain Pathology, 19, 214-223. http://dx.doi.org/10.1111/j.1750-3639.2008.00176.x [63] Mongiat, L.A. and Schinder, A.F. (2011) Adult Neurogenesis and the Plasticity of the Dentate Gyrus Network. European Journal of Neuroscience, 33, 1055-1061. http://dx.doi.org/10.1111/j.1460-9568.2011.07603.x [64] Lee, J.W., Lee, Y.K., Yuk, D.Y., Choi, D.Y., Ban, S.B., Oh, K.W. and Hong, J.T. (2008) Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment through Enhancement of Beta-Amyloid Generation. Journal of Neuroinflammation, 5, 37. http://dx.doi.org/10.1186/1742-2094-5-37 [65] Sheng, J.G., Bora, S.H., Xu, G., Borchelt, D.R., Price, D.L. and Koliatsos, V.E. (2003) Lipopolysaccharide-InducedNeuroinflammation Increases Intracellular Accumulation of Amyloid Precursor Protein and Amyloid β Peptide in APPswe Transgenic Mice. Neurobiology of Disease, 14, 133-145. http://dx.doi.org/10.1016/S0969-9961(03)00069-X [66] Joshi, Y.B., Giannopoulos, P.F., Chu, J. and Praticò, D. (2014) Modulation of Lipopolysaccharide-Induced Memory Insult, γ-Secretase, and Neuroinflammation in Triple Transgenic Mice by 5-Lipoxygenase. Neurobiology of Aging, 35, 1024-1031. http://dx.doi.org/10.1016/j.neurobiolaging.2013.11.016 [67] Qiao, X., Cummins, D.J. and Paul, S.M. (2001) Neuroinflammation-Induced Acceleration of Amyloid Deposition in the APPV717F Transgenic Mouse. European Journal of Neuroscience, 14, 474-482. http://dx.doi.org/10.1046/j.0953-816x.2001.01666.x [68] DiCarlo, G., Wilcock, D., Henderson, D., Gordon, M. and Morgan, D. (2001) Intrahippocampal LPS Injections Reduce Aβ Load in APP+PS1 Transgenic Mice. Neurobiology of Aging, 22, 1007-1012. http://dx.doi.org/10.1016/S0197-4580(01)00292-5 [69] Herber, D.L., Mercer, M., Roth, L.M., Symmonds, K., Maloney, J., Wilson, N., Freeman, M.J., Morgan, D. and Gordon, M.N. (2007) Microglial Activation Is Required for Aβ Clearance after Intracranial Injection of Lipopolysaccharide in APP Transgenic Mice. Journal of Neuroimmune Pharmacology, 2, 222-231. http://dx.doi.org/10.1007/s11481-007-9069-z [70] Kandalepas, P.C., Sadleir, K.R., Eimer, W.A., Zhao, J., Nicholson, D.A. and Vassar, R. (2013) The Alzheimer’s β-Secretase BACE1 Localizes to Normal Presynaptic Terminals and to Dystrophic Presynaptic Terminals Surrounding Amyloid Plaques. Acta Neuropathologica, 126, 329-352. http://dx.doi.org/10.1007/s00401-013-1152-3 Scientific Research Publishing (SCIRP) is one of the largest Open Access journal publishers. It is currently publishing more than 200 open access, online, peer-reviewed journals cov







论文发表 是一个专门从事期刊推广、论文发表、论文发表辅导的机构。融合收集数百家期刊杂志社征稿评职称,发表论文,以供广大作者免费阅览,以期待各位作者在短时间内掌握每种期刊征稿要求、审稿范围,在日常繁重的工作中短时间开心、放心快速投稿、在适合的时间拿到刊物,评上职称,不为挤不上评职称的末班车而烦恼。
本站主要整合如下评职称,发表论文:
教育论文发表
经济论文发表
科技论文发表
医学论文发表
计算机论文发表
文学论文发表
农业论文发表
学报论文发表
其它论文发表等,另期待更多的杂志编辑与本站联系文:
85782530(普刊发表)
82534308(核心发表)共同以正期刊界正刊之气,维护学术良好的氛围。
免责声明:本网所提供的信息资源如有侵权、违规,请及时告知!
论文网版权所有, 本站提供论文发表 论文投稿 发表论文 发表文章
文章只代表作者观点,并不意味着本站认同,部分作品系转载,版权归原作者或相应的机构;若某篇作品侵犯您的权利,请来信告知:lunww@126.com
中国论文网全权所属国际域名:www.lunww.com 中文域名:论文发表 技术支持:论文发表 国家双软认定单位 法律顾问:全民安律师事务所
期刊合作加盟咨询:5715378(加盟、投诉、建议) 论文发表咨询电话:18262951856 论文发表投稿邮箱:lunww@126.com
Powered by 论文发表 © 2010-2020

苏ICP备19023845号-4